本篇文章为大家展示了KEGGgraph怎样根据kgml 文件从pathway中重构出基因互作网络,内容简明扼要并且容易理解,绝对能使你眼前一亮,通过这篇文章的详细介绍希望你能有所收获。
KEGGgraph 包可以解析kgml 文件,从中得到不同对象之间的网络结构,并在此基础上进一步挖掘其中的信息。
KEGGgraph 包提供了以下3种基本功能:
用法示例
# 读取hsa00020xml 文件
> mapkG <- parseKGML2Graph("hsa00020.xml",expandGenes=TRUE, genesOnly = TRUE)
> mapkG
A graphNEL graph with directed edges
Number of Nodes = 30
Number of Edges = 101
> nodes(mapkG)
[1] "hsa:1738" "hsa:4967" "hsa:55753" "hsa:1743" "hsa:8801" "hsa:8802"
[7] "hsa:8803" "hsa:3417" "hsa:3418" "hsa:3419" "hsa:3420" "hsa:3421"
[13] "hsa:47" "hsa:2271" "hsa:48" "hsa:50" "hsa:1431" "hsa:4190"
[19] "hsa:4191" "hsa:5091" "hsa:5160" "hsa:5161" "hsa:5162" "hsa:1737"
[25] "hsa:5105" "hsa:5106" "hsa:6389" "hsa:6390" "hsa:6391" "hsa:6392"
> edges(mapkG)
$`hsa:1738`
[1] "hsa:4967" "hsa:55753" "hsa:5160" "hsa:5161" "hsa:5162" "hsa:1737"
$`hsa:4967`
[1] "hsa:3419" "hsa:3420" "hsa:3421" "hsa:3417" "hsa:3418"
$`hsa:55753`
[1] "hsa:3419" "hsa:3420" "hsa:3421" "hsa:3417" "hsa:3418"在 parseKGML2Graph 中,有两个参数,expandGenes 和 genesOnly。
expandGenes 控制是否将基因进行展开,在pathway 中,会有1个KO 对应多个gene的情况,比如下面这种
<entry id="32" name="hsa:8801 hsa:8802 hsa:8803" type="gene" reaction="rn:R00405"
link="">http://www.kegg.jp/dbget-bin/www_bget?hsa:8801+hsa:8802+hsa:8803">;
<graphics name="SUCLG2, G-SCS, GBETA, GTPSCS..." fgcolor="#000000" bgcolor="#BFFFBF"
type="rectangle" x="260" y="574" width="46" height="17"/>
</entry>expandGenes = TRUE 表示将基因展开,每个基因作为一个节点。
genesOnly 参数控制是否将其他类型的entry (比如compound等类型)展现在network 中,默认值为 TRUE,所以最终得到的network 中节点全部是基因。
通过parseKGML2Graph 这一步我们就可以从一张pathway 中得到基因产物(蛋白)的互作网络, 还需要注意一点,整个网络是一个有向图, 因为基因产物之间的互作关系是由方向性的。
由于自带的可视化不够美观,我们把nodes和edges 写入文件,用cytoscape 进行可视化,用法示例
mapkNodes <- nodes(mapkG)
mapkEdges <- edges(mapkG)
mapkEdges <- mapkEdges[sapply(mapkEdges, length) > 0]
res <- lapply(1:length(mapkEdges), function(t){
name <- names(mapkEdges)[t]
len <- length(mapkEdges[[t]])
do.call(rbind, lapply(1:len, function(n){
c(name, mapkEdges[[t]][n])
}))
})
result <- data.frame(do.call(rbind, res))
write.table(result, "edges.txt", sep = "\t", row.names = F, col.names = F, quote = F)
write.table(mapkNodes, "nodes.txt", sep = "\t", row.names = F, col.names = F, quote = F)导入cytoscape 画出来的图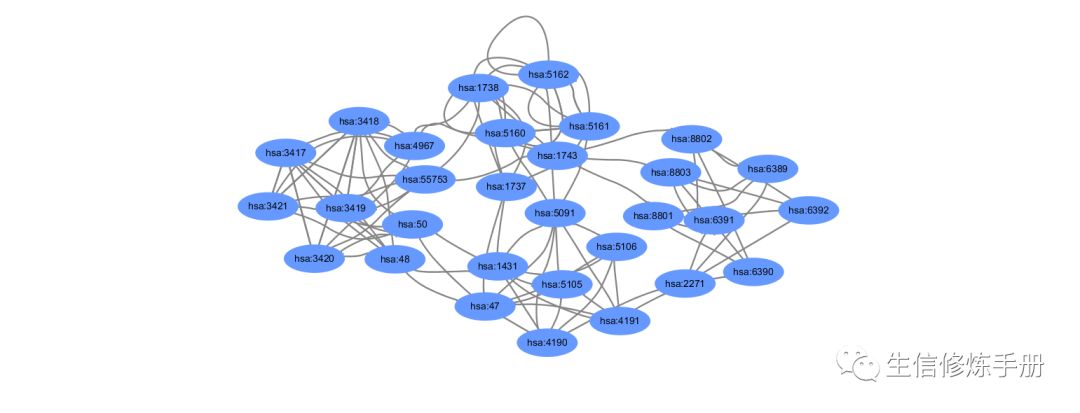
degree信息对于一个netwrok 而言,每个节点的degree 信息是我们最常用的信息, 示例
> mapkGoutdegrees <- sapply(edges(mapkG), length)
> mapkGindegrees <- sapply(inEdges(mapkG), length)
> degrees <- data.frame(indegrees = mapkGindegrees, outdegrees = mapkGoutdegrees)
> head(degrees)
indegrees outdegrees
hsa:1738 1 6
hsa:4967 2 5
hsa:55753 2 5
hsa:1743 3 3
hsa:8801 4 1
hsa:8802 4 1由于是有向图,所以有入度 indegrees 和 出度 outdegrees 的概念。
除了以上基础功能外,还可以借助其他的R包进一步挖掘信息,比如在整个基因互作网络, 哪个基因是最关键的。
示例:
> library(RBGL)
> mapkG <- parseKGML2Graph("hsa00020.xml",expandGenes=TRUE, genesOnly = TRUE)
> bcc <- brandes.betweenness.centrality(mapkG)
> rbccs <- bcc$relative.betweenness.centrality.vertices[1L,]
> toprbccs <- sort(rbccs,decreasing=TRUE)[1:4]
> toprbccs
hsa:1743 hsa:2271 hsa:1738 hsa:47
0.21597893 0.16177167 0.14965648 0.09880362对于network 而言,我们一般认为degree 越大的点在这个网络中越重要,所以需要看节点的degree 信息。除了这种基本的认识外,还有很多成熟的算法,从network 中挖掘关键节点。 RBGL 包提供了Brandes 的算法,用来衡量节点在网络中的重要性,上面的结果中,toprbccs 就是我们筛选出的4个比较重要的节点。
使用KEGGgraph包,我们可以方便的从pathway中得到基因户做网络;
可以将network 中的nodes和edges 信息导出,使用cytoscape 可视化;
可以借助其他成熟的算法挖掘基因互作网络中的关键基因;
上述内容就是KEGGgraph怎样根据kgml 文件从pathway中重构出基因互作网络,你们学到知识或技能了吗?如果还想学到更多技能或者丰富自己的知识储备,欢迎关注亿速云行业资讯频道。
亿速云「云服务器」,即开即用、新一代英特尔至强铂金CPU、三副本存储NVMe SSD云盘,价格低至29元/月。点击查看>>
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。
原文链接:https://my.oschina.net/u/4580290/blog/4626681



 计算
计算 安全
安全 数据库
数据库 网络和加速
网络和加速 企业服务
企业服务



