这篇文章主要讲解了“如何从FASTQ转换得到uBAM格式”,文中的讲解内容简单清晰,易于学习与理解,下面请大家跟着小编的思路慢慢深入,一起来研究和学习“如何从FASTQ转换得到uBAM格式”吧!
二代测序平台产生的数据通常用fastq格式进行存储,fastq 存储了我们最关心的序列和碱基质量的信息。就测序而言,这样的信息当然是足够了。但是对于分析而言,还缺少了一点信息。
给你一个fastq文件,你最多可以看出来样本名,测序平台,测序读长等基本信息,如果想知道测序类型(是WES, WGS 还是RNA-seq), 样本的采样信息,样本的分组信息,这些信息从fastq 文件是无法得到的。这些实验相关的数据,称之为metadata。
uBAM和FASTQ相比,处理存储了序列和碱基质量信息之外,还可以存储metadata信息。
GATK4中,数据预处理部分的示意图如下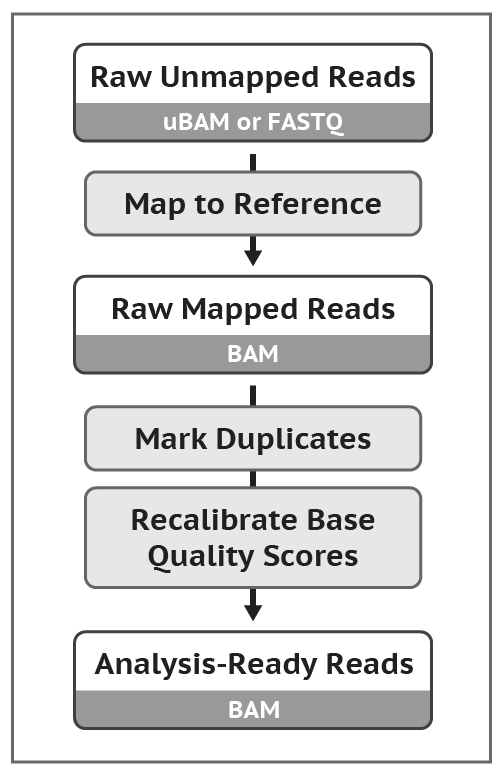
可以看到,对于原始数据,有两种格式,一种就是我们常见的FASTQ; 另外一种就是uBAM。官方更加推荐使用uBAM格式。
如何从FASTQ转换得到uBAM格式呢?我们需要借助picatd工具。picard提供了一个FastqToSam功能,可以将序列转换成ubam格式。
基本用法如下:
java -jar picard.jar FastqToSam
F1=sampleA_R1.fastq.gz
F2=sampleA_R2.fastq.gz
PL=illumina
SM=sampleA
LB=sampleA
RG=sampleA
O=sampleA.ubam
F1和F2指定原始的fastq格式的数据,对于双端测序,同时指定F1和F2, 对于单端测序,指定F1就可以了。PL代表platform, 指定测序平台,取值包含 illumina 和 solid 两种;SM代表 sample name, 指定样本名称;LB代表library name, 指定文库名称,RG代表read group, 指定reads group的名字,这两个参数一般和样本名相同就可以了。
ubam从名称上也可以看出来,是属于bam格式的,所以其内容也分成了头部和正文两个部分。
samtools view -H sampleA.ubam
@HD VN:1.5 SO:queryname
@RG ID:sampleA SM:sampleA LB:sampleA PL:illumina
第一行是标准的bam文件头部的声明,第二行的@RG就是转换过程中添加的几种metadata信息。
samtools view sampleA.ubam
由于列数较多,这里我截取了前面几列
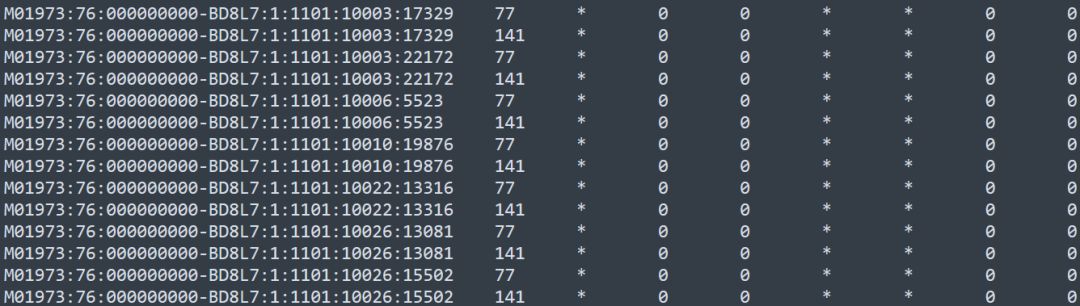
每一行代表一条序列,序列ID相同的实际上是R1和R2端,从第二列的flag可以区分R1和R2端。
samtools flags 77
0x4d 77 PAIRED,UNMAP,MUNMAP,READ1
samtools flags 141
0x8d 141 PAIRED,UNMAP,MUNMAP,READ2
77对应R1端, 141对应R2端。
第三列的*代表没有比对上染色体,这就是unmapped bam的由来。
通过FastqToSam可以从fastq文件得到ubam文件,picard 还提供了SamtoFastq命令,从bam 文件得到fastq 文件
用法如下:
java -jar picard.jar SamToFastq
I=sampleA.ubam
F=sampleA_R1.fastq
F2=sampleA_R2.fastq
I代表input, 指定输入的bam 文件;F和F2 指定输出的fastq 文件。
感谢各位的阅读,以上就是“如何从FASTQ转换得到uBAM格式”的内容了,经过本文的学习后,相信大家对如何从FASTQ转换得到uBAM格式这一问题有了更深刻的体会,具体使用情况还需要大家实践验证。这里是亿速云,小编将为大家推送更多相关知识点的文章,欢迎关注!
亿速云「云服务器」,即开即用、新一代英特尔至强铂金CPU、三副本存储NVMe SSD云盘,价格低至29元/月。点击查看>>
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。
原文链接:https://my.oschina.net/u/4580290/blog/4626602



 计算
计算 安全
安全 数据库
数据库 网络和加速
网络和加速 企业服务
企业服务



