本篇内容介绍了“kallisto怎么使用”的有关知识,在实际案例的操作过程中,不少人都会遇到这样的困境,接下来就让小编带领大家学习一下如何处理这些情况吧!希望大家仔细阅读,能够学有所成!
kallisto 是2016年发布的一款无须比对的转录本定量工具,采用了名为pseudo-alignment的算法。传统的定量算法是根据reads的比对位置来确认其属于哪个转录本或者基因,而pseudo-alignment 算法不关系reads具体的比对位置,而是通过reads的kmer特征来判断其属于哪一条转录本,示意图如下
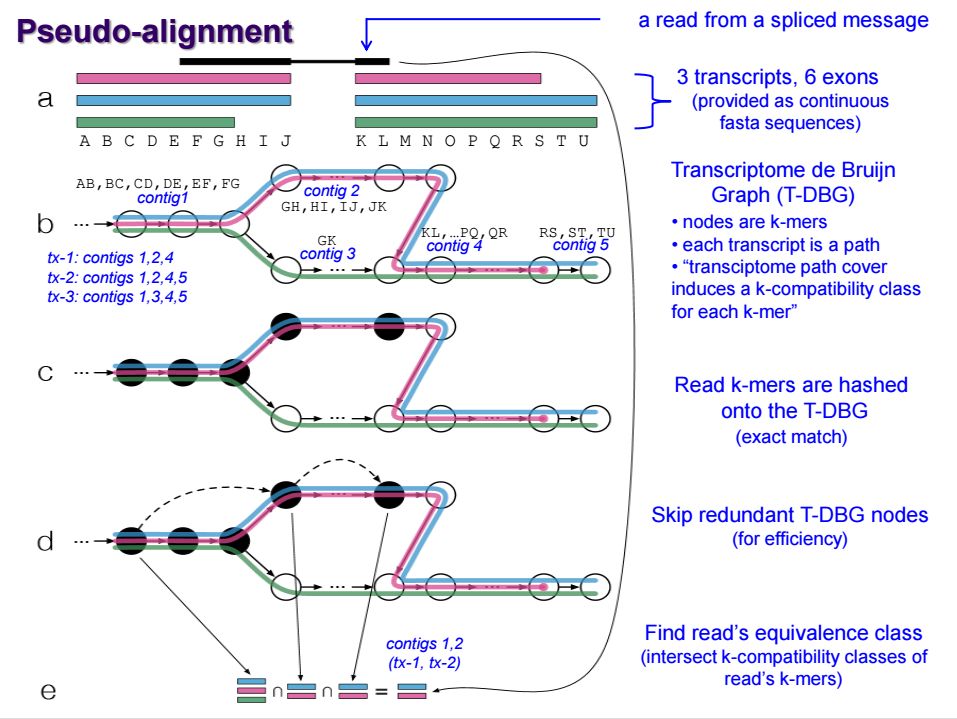
首先将每个转录本序列划分为kmer, 利用所有转录本的kmer序列构建de Bgujin Graph, 简称T-DBG,在这个图中,每个节点是一个kmer, 每条路径代表一个转录本, 由于转录本序列的冗余,实际上每个kmer对应多条路径,也就是对应多个转录本; 然后将测序的reads也划分为kmer, 并将其映射到T-DBG中。
最终定量时,将该reads的所有kmer对应的转录本取交集,就能够分析出reads可能属于哪些转录本序列。
官网有编译好的可执行文件,下载解压即可。代码如下
wget https://github.com/pachterlab/kallisto/releases/download/v0.44.0/kallisto_linux-v0.44.0.tar.gz tar xzvf kallisto_linux-v0.44.0.tar.gz
解压之后,在文件夹下可以看到名为kallisto的可执行文件。从算法也可以看到,软件的运行需要两步,第一步对转录本的序列划分kmer, 构建T-DBG, 也称之为建索引;第二步对reads 定量。
kallisto支持读取gzip压缩的转录本序列,用法如下
kallisto index -k 31 -i hg19.idx hg19.refMrna.fa
只需要提供转录本的fasta格式的序列即可。-k参数指定kmer的长度,-i参数指定输出的索引的名字,注意kallisto建立的索引为一个文件。
kallisto 支持单端和双端数据的定量,双端数据用法如下
kallisto quant \ -i hg19.idx \ -o out_dir \ -t 20 \ R1.fastq.gz R2.fastq.gz
-i参数指定转录本的索引文件,-o参数指定输出结果的目录,-t参数指定线程数,kallisto支持gzip压缩的序列文件。
单端数据用法如下
kallisto quant \ -i hg19.idx \ -o output \ --single \ -l 180 \ -s 20 \ -t 20 \ reads.fastq.gz
对于单端数据而言,必须指定fragment长度的均值和方差,分别对应-l和-s参数。
在输出目录,会生成以下3个文件
├── abundance.h6 ├── abundance.tsv └── run_info.json
run_info.json 文件为JSON格式,保存了运行的命令和参数。
前缀为abundance 的文件,保存了转录本的定量信息。其中h6为HDF5格式的文件,当转录本数量较多时,相比纯文本,这种格式的文件大小会小很多;tsv为纯文本的文件,内容如下
| target_id | length | eff_length | est_counts | tpm |
|---|---|---|---|---|
| NR_103451 | 865 | 664.449 | 9 | 0.493026 |
| NM_001243523 | 577 | 376.636 | 31 | 2.99591 |
| NR_038931 | 2432 | 2231.4 | 36.9964 | 0.603491 |
对于HDF5的文件,可以采用如下命令转换为tsv格式的文件
kallisto h6dump -o out_dir abundance.h6
-o参数指定输出结果的目录,最终生成的文件名称为abundance.tsv。
“kallisto怎么使用”的内容就介绍到这里了,感谢大家的阅读。如果想了解更多行业相关的知识可以关注亿速云网站,小编将为大家输出更多高质量的实用文章!
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。





