怎样使用Mfuzz进行时间序列表达模式聚类分析,很多新手对此不是很清楚,为了帮助大家解决这个难题,下面小编将为大家详细讲解,有这方面需求的人可以来学习下,希望你能有所收获。
在之前的文章中,我们介绍了STEM软件,针对时间序列的数据,可以进行基因表达模式聚类分析。这里介绍另外一个功能相同的R包Mfuzz。这个R包的地址如下
https://www.bioconductor.org/packages/release/bioc/html/Mfuzz.html
Mfuzz采用了一种新的聚类算法fuzzy c-means algorithm,在文献中称这种聚类算法为soft clustering算法,相比K-means等hard clustering算法,一定程度上降低了噪声对聚类结果的干扰,而且这种算法有效的定义了基因和cluster之间的关系,即基因是否属于某个cluster, 对应的值为memebership。
对于分析而言,我们只需要提供基因表达量的数据就可以了,需要注意的是,Mfuzz默认你提供的数据是归一化之后的表达量,这意味着表达量必须可以直接在样本间进行比较,对于FPKM, TPM这两种定量方式而言,是可以直接在样本间进行比较的,但是对于count的定量结果,我们必须先进行归一化,可以使用edgeR或者DESeq先得到归一化之后的数据在进行后续分析。
假设你已经有归一化之后的表达量了,通过以下几个步骤就可以得到聚类的结果
预处理包括读取数据,去除表达量太低或者在不同时间点间变化太小的基因等步骤,代码如下
x <- read.table(
"normalisation.count.txt",
row.names = 1,
sep = "\t",
header = T)
count <- data.matrix(x)
eset <- new("ExpressionSet",exprs = count)
# 根据标准差去除样本间差异太小的基因
eset <- filter.std(eset,min.std=0)需要注意的是,Mfuzz聚类时要求是一个ExpressionSet类型的对象,所以需要先用表达量构建这样一个对象。
聚类时需要用一个数值来表征不同基因间的距离,Mfuzz中采用的是欧式距离,由于普通欧式距离的定义没有考虑不同维度间量纲的不同,所以需要先进行标准化,代码如下
eset <- standardise(eset)Mfuzz中的聚类算法需要提供两个参数,第一个参数为希望最终得到的聚类的个数,这个参数由我们直接指定;第二个参数称之为fuzzifier值,用小写字母m表示,可以通过函数评估一个最佳取值,代码如下
# 聚类个数
c <- 6
# 评估出最佳的m值
m <- mestimate(eSet)
# 聚类
cl <- mfuzz(eSet, c = c, m = m)在cl这个对象中就保存了聚类的完整结果,对于这个对象的常见操作如下
# 查看每个cluster中的基因个数
cl$size
# 提取某个cluster下的基因
cl$cluster[cl$cluster == 1]
# 查看基因和cluster之间的membership
cl$membership代码如下
mfuzz.plot(
eSet,
cl,
mfrow=c(2,3),
new.window= FALSE)生成的图片如下
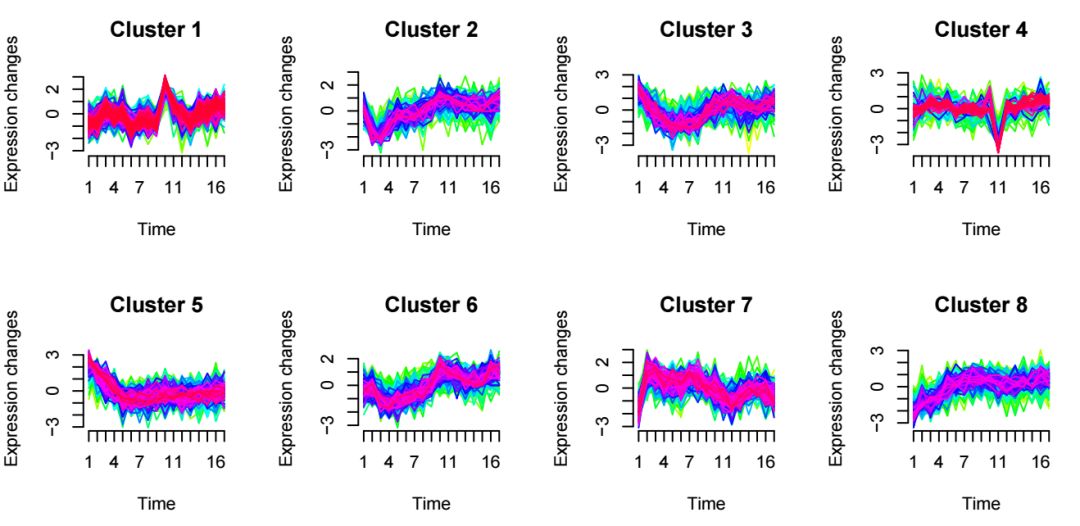
对于感兴趣的表达模式,可以用上述提到的用法提取出该cluster下的基因列表,通过GO/KEGG等功能富集分析进行深入挖掘。
看完上述内容是否对您有帮助呢?如果还想对相关知识有进一步的了解或阅读更多相关文章,请关注亿速云行业资讯频道,感谢您对亿速云的支持。
亿速云「云服务器」,即开即用、新一代英特尔至强铂金CPU、三副本存储NVMe SSD云盘,价格低至29元/月。点击查看>>
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。
原文链接:https://my.oschina.net/u/4580290/blog/4620554



 计算
计算 安全
安全 数据库
数据库 网络和加速
网络和加速 企业服务
企业服务



