今天就跟大家聊聊有关find_circ中如何识别环状RNA,可能很多人都不太了解,为了让大家更加了解,小编给大家总结了以下内容,希望大家根据这篇文章可以有所收获。
官方的pipeline使用的是bowtie2软件,代码如下
bowtie2 -p16 \
--very-sensitive \
--score-min=C,-15,0 \
--mm \
-x hg19 -q \
-1 R1.fastq.gz -2 R2.fastq.gz \
2> bowtie2.log \
| samtools view -hbuS - \
| samtools sort - accepted_hits最终生成了一个排序之后的bam文件,其实这一步选择其他的比对软件,比如hisat也是可以的,只需要产生bam文件就可以了。
采用samtools软件提取没比对上的序列,代码如下
samtools view -hf 4 accepted_hits.bam | samtools view -Sb - > unmapped.bam代码如下
unmapped2anchors.py unmapped.bam anchor.fq
bowtie2 -p 16 \
--reorder \
--mm \
--score-min=C,-15,0 \
-q -x human_bowtie2_index \
-U anchor.fq \
-S align.sam代码如下
cat align.sam | find_circ.py -G hg19.fa -p hsa_ > splice_sites.bed结果如下所示
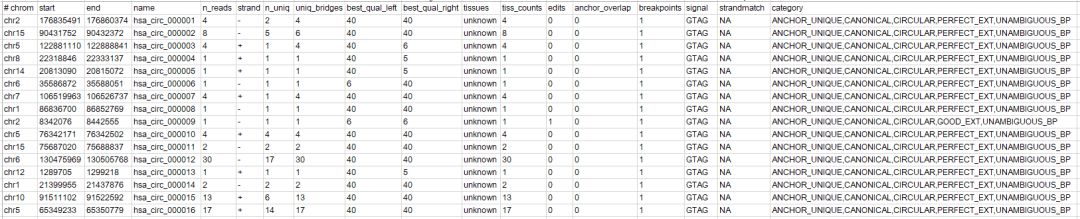
-p参数指定的是第四列内容的前缀,建议指定为物种对应的三字母缩写,需要注意的是,在sites.bed中同时包含了环状RNA和线性RNA,环状RNA的名称用circ标识,线性RNA的名称用norm标识。
根据以下规则对结果进行筛选
根据关键词CIRCULAR筛选环状RNA
去除线粒体上的环状RNA
筛选unique junction reads数至少为2的环状RNA
去除断裂点不明确的环状RNA
过滤掉长度大于100kb的circRNA,这里的100kb为基因组长度,直接用环状RNA的头尾相减即可
代码如下
grep CIRCULAR splice_sites.bed | \
grep -v chrM | \
awk '$5>=2' | \
grep UNAMBIGUOUS_BP | \
grep ANCHOR_UNIQUE | \
./maxlength.py 100000 \
> circ_candidates.bed看完上述内容,你们对find_circ中如何识别环状RNA有进一步的了解吗?如果还想了解更多知识或者相关内容,请关注亿速云行业资讯频道,感谢大家的支持。
亿速云「云服务器」,即开即用、新一代英特尔至强铂金CPU、三副本存储NVMe SSD云盘,价格低至29元/月。点击查看>>
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。
原文链接:https://my.oschina.net/u/4580290/blog/4602559



 计算
计算 安全
安全 数据库
数据库 网络和加速
网络和加速 企业服务
企业服务



