今天就跟大家聊聊有关如何理解CGA中的分析结果,可能很多人都不太了解,为了让大家更加了解,小编给大家总结了以下内容,希望大家根据这篇文章可以有所收获。
TCGA对于不同类型的数据,有着独特的处理流程,具体如下
TCGA中的DNA测序主要用来分析肿瘤患者中的体细胞突变,和GATK的体细胞突变流程类似,前期都经过了一个预处理步骤,这里称之为co-cleanning, 流程示意如下
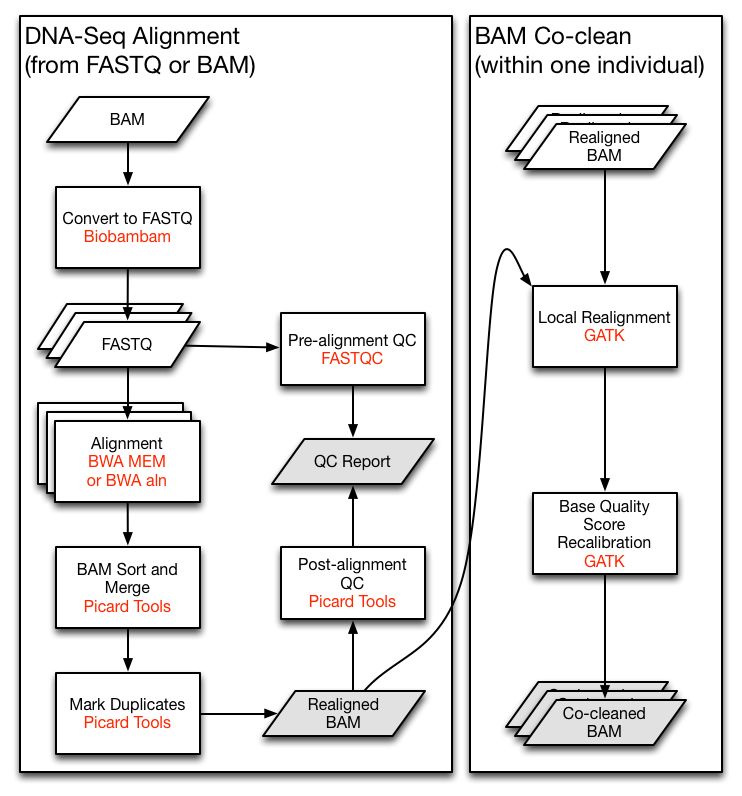
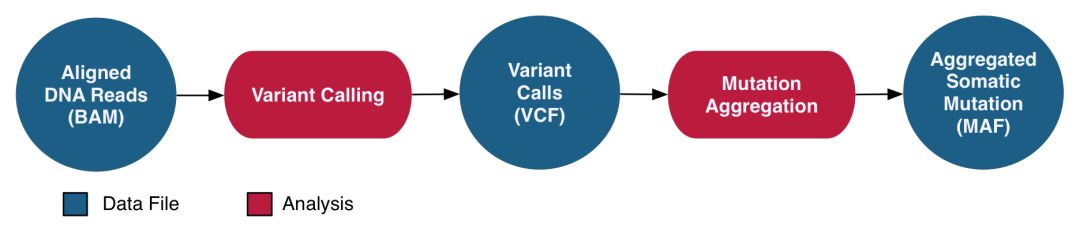
就是经典的sort->markduplicate->Realign->BQSR步骤,得到co-cleaned BAM文件。然后用配对的肿瘤和正常样本进行somatic variant calling, 得到VCF文件。然后进行体细胞突变的注释,得到突变注释文件MAF, 示意如下
在进行体细胞突变位点分析时,使用了以下4款不同的软件同时分析
MuSE
Mutect2
SomaticSniper
Varscan2
各自对应的pipeline示意如下
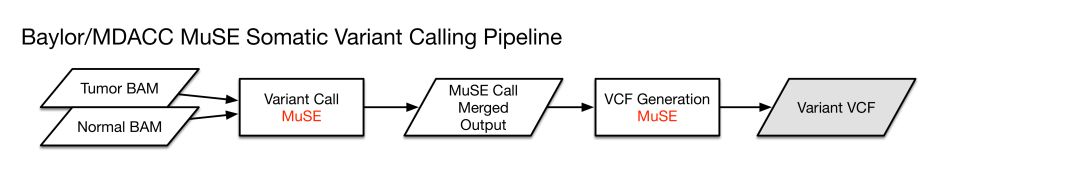
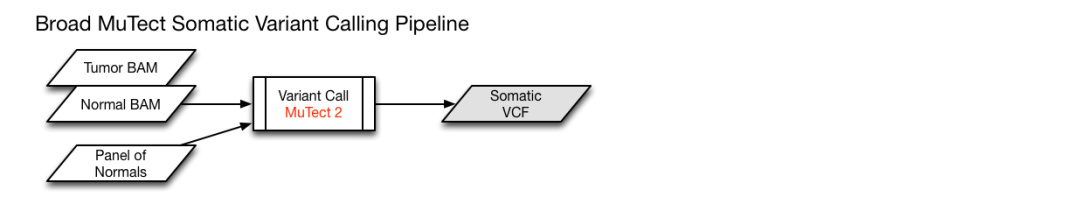
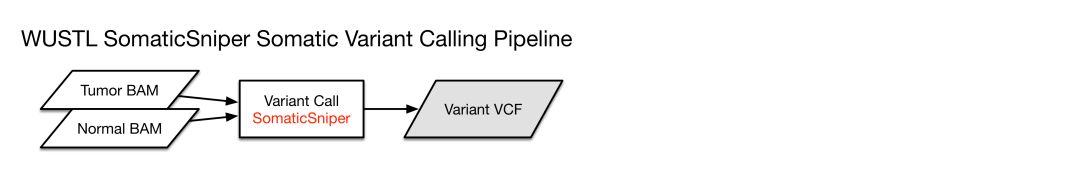
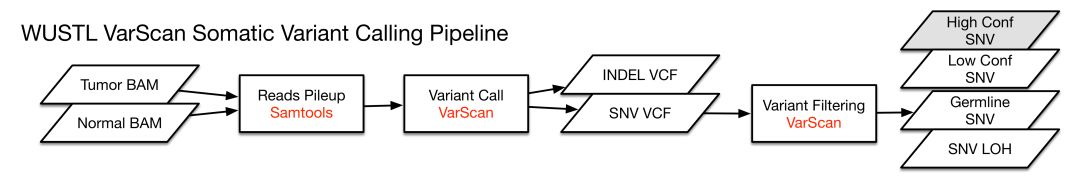
各自pipeline得到的VCF文件,使用VEP软件对体细胞突变位点进行注释,使用了以下数据库进行注释
GENCODE v.22
sift v.5.2.2
ESP v.20141103
polyphen v.2.2.2
dbSNP v.146
Ensembl genebuild v.2014-07
Ensembl regbuild v.13.0
HGMD public v.20154
ClinVar v.201601
注释完成之后,会对突变位点进行过滤,去除低质量的突变位点和潜在的生殖细胞突变位点,剩余的位点作为最终的体细胞突变位点,保存在MAF文件中供下载。
当然对于没有配对的正常样本,也有tumor-only variant calling workflow来处理,具体请参考以下链接
https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/DNA_Seq_Variant_Calling_Pipeline
mRNA分析是通过STAR的2-pass模式比对hg38参考基因组,然后使用HTSeq进行定量,定量时基于Gencode V22版本的GTF文件,流程示意如下
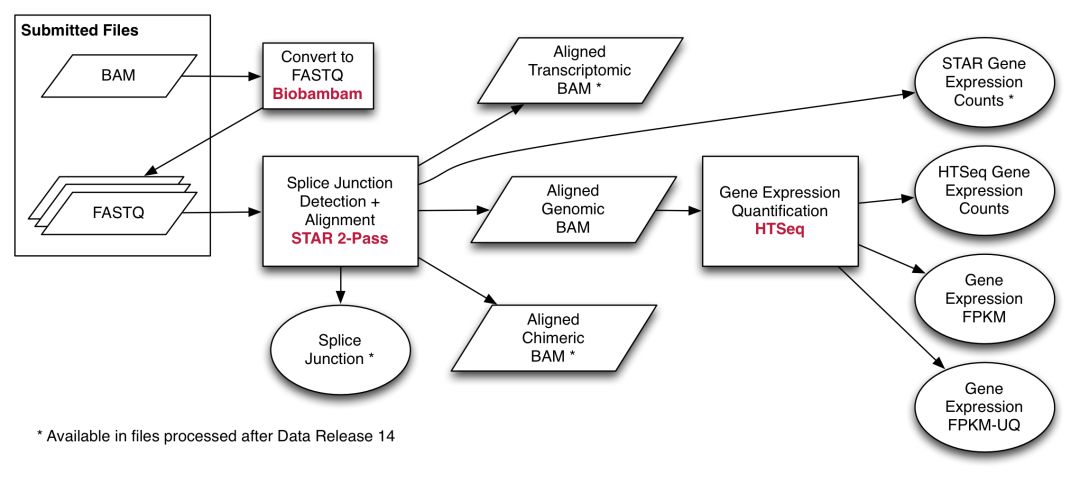
在定量时,提供了以下3种策略
Raw count
FPKM
FPKM-UQ
Raw count和FPKM是转录组分析中经典的定量策略,而FPKM-UQ则是在FPKM基础上新提出的一种策略,计算公式如下
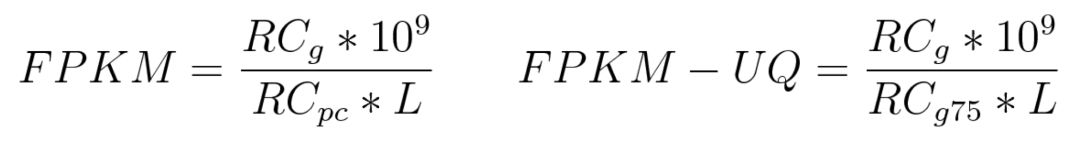
和FPKM不同的是,在FPKM-UQ中采用所有基因Mapping reads数目的上四分位数代替了所有基因Mapping Reads的总数。官方也提供了一个示例帮助我们理解具体的计算过程

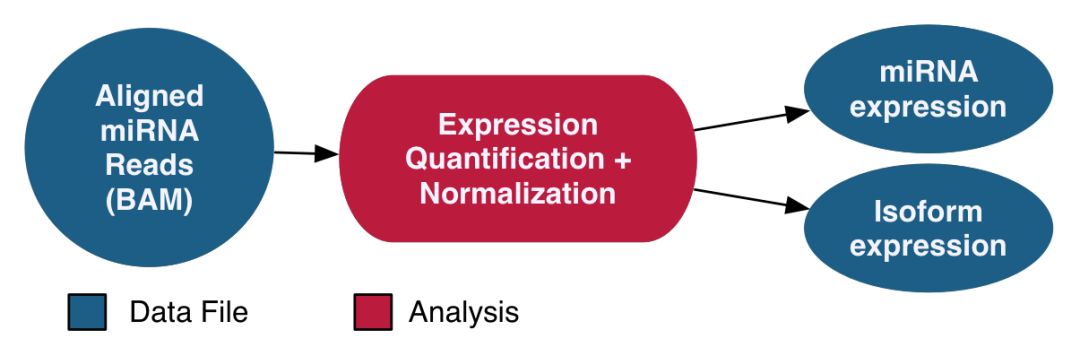
miRNA的分析采用了BCGSC开发的miRNA定量流程,这套流程只针对已知的miRNA进行定量,链接如下
https://github.com/bcgsc/mirna
流程示意如下
使用Affymetrix SNP 6.0芯片来分析CNV, 首先使用DNACopy这个R包来计算拷贝数,然后用GISTIC2根据CNV来评估基因的变化情况,是loss还是gain, 流程示意如下

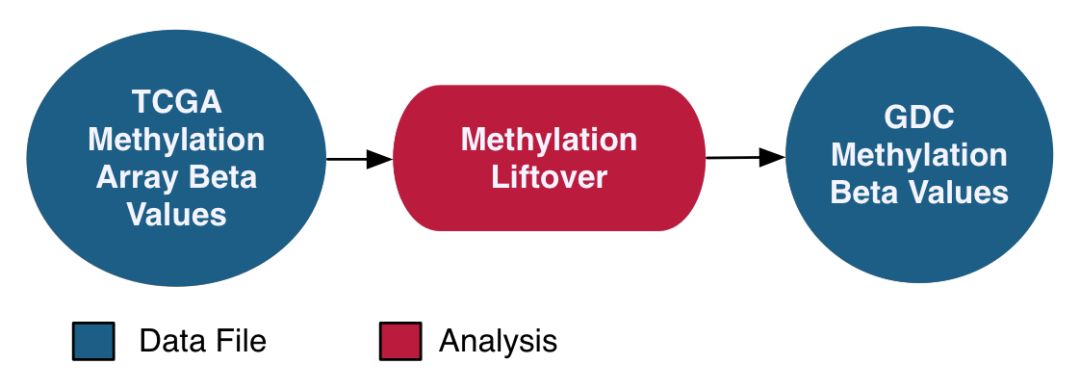
通过illumina Infinum Human Methylation 27和HumanMethylation450 两个芯片平台来分析DNA甲基化,采用了beta值的定量策略。同时考虑到这两个探针是针对hg19来设计的,将探针序列与hg38进行比对,当MAPQ<10或者I型和II型探针比对到不同基因组区域时,过滤到这部分探针。剩余的CpG文件根据GENCODE V22版本的GTF来进行注释,根据这样的策略将hg19上的甲基化移植到hg38版本的基因组上,具体流程示意如下
了解TCGA数据分析的流程,可以更好的在GDC数据库中筛选数据,也可以更好的和自己的数据进行比较。
看完上述内容,你们对如何理解CGA中的分析结果有进一步的了解吗?如果还想了解更多知识或者相关内容,请关注亿速云行业资讯频道,感谢大家的支持。
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。





