本篇内容主要讲解“fastANI怎么用”,感兴趣的朋友不妨来看看。本文介绍的方法操作简单快捷,实用性强。下面就让小编来带大家学习“fastANI怎么用”吧!
在比较基因组分析中,我们经常需要分析不同基因组之间的进化关系,例如我们可以使用标记蛋白来构建系统发育树。为了进行定量的比较,我们还可以计算不同基因组之间的相似性或者进化距离,以进行物种分类、亲缘关系比较等。平均核苷酸相似度(Average Nucleotide Identity,ANI)是在核苷酸水平比较两个基因组亲缘关系的指标。ANI被定义为两个微生物基因组同源片段之间平均的碱基相似度,他的特点是在近缘物种之间有较高的区分度。
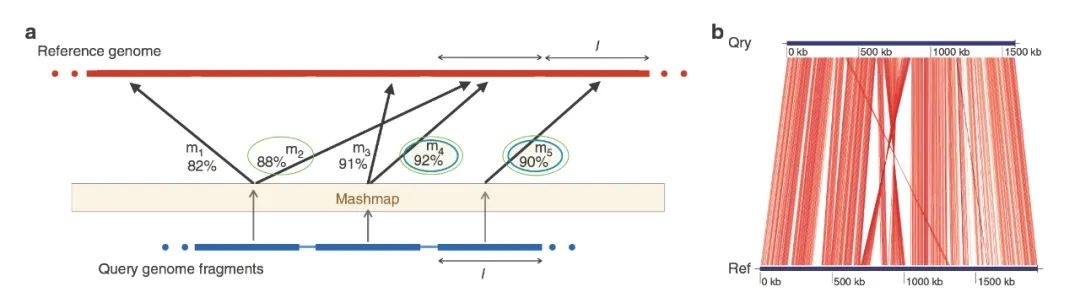
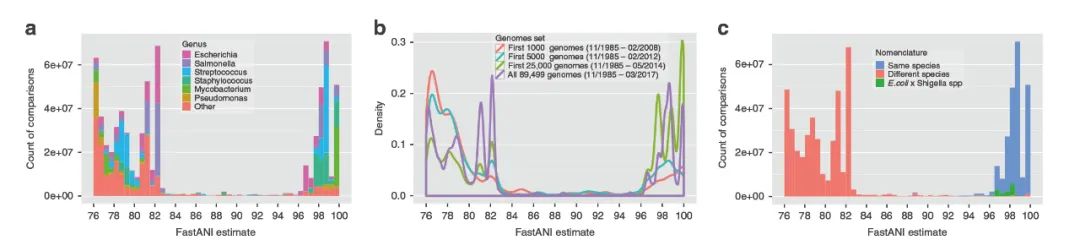
在最近Nature communications的一篇研究中,作者使用fastANI对9万个基因组进行分析,发现大多数谱系种内与种间存在一个明显的ANI分界线,相同物种的基因组ANI小于95%,不同物种的基因组ANI大于95%,因此常以95%的ANI作为物种划分与物种聚类的标准[1]。
fastANI从GitHub下载软件包解压就可以使用,其使用方法如下所示:
fastANI -q genome1.fa -r genome2.fa -o output.txtfastANI -q genome1.fa --rl genome_list.txt -o output.txt-r, --ref:参考基因组核苷酸序列,可以试fasta/fastq及其gzip压缩文件--rl, --refList:包含参考基因组列表的文件,从而允许多个参考基因组-q, --query:查询基因组核苷酸序列,可以试fasta/fastq及其gzip压缩文件--ql, --queryList:包含查询基因组列表的文件,从而允许多个查询基因组-k, --kmer:比对的kmer大小,不能大于16,默认为16-t, --threads:程序运行所使用的核数,默认为1--fragLen:片段长度,默认为3000--minFrag:最短匹配的片段,默认为50--visualize:输出比对图像,只适用于一对一比对,默认关闭--matrix:输出ANI值作为下三角矩阵,适用于多对多比对,默认关闭-o, --output:输出文件名
fastANI -q 951_armatimo.fasta -r 391_armatimo.fasta -o output1.txt --fragLen 1000
结果如下所示:

其ANI为74.7,2570为参考基因组的所有序列片段,981为查询基因组中比对上的同源片段,片段数过少的ANI值是没有意义的,可以去掉。
多个基因组互相比较如下所示:
fastANI --ql Armatimonadetes.txt --rl Armatimonadetes.txt -o output2.txt --fragLen 1000 -t 10 --matrix
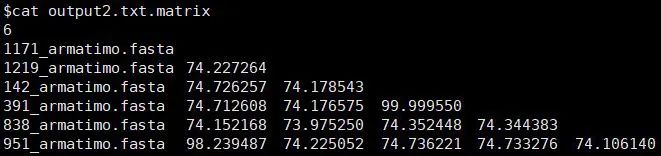
生成的矩阵结果如下所示:
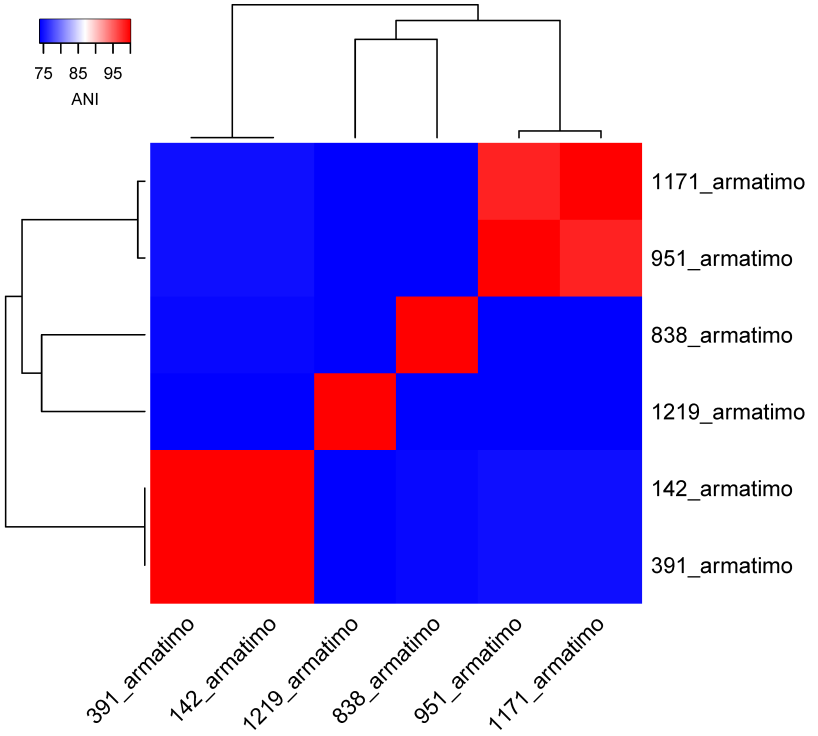
以上矩阵我们可以在R中作图展示,如下所示:
到此,相信大家对“fastANI怎么用”有了更深的了解,不妨来实际操作一番吧!这里是亿速云网站,更多相关内容可以进入相关频道进行查询,关注我们,继续学习!
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。





