今天小编给大家分享一下怎么使用python服务器批处理得到PSSM矩阵的相关知识点,内容详细,逻辑清晰,相信大部分人都还太了解这方面的知识,所以分享这篇文章给大家参考一下,希望大家阅读完这篇文章后有所收获,下面我们一起来了解一下吧。
最好新建一个python环境,因为我发现conda安装blast默认的是python==3.6.11,可能会不小心把你的python版本改掉…然后你写好的代码全die了……
conda create -n blast python==3.6.11
source activate blast
conda install -c bioconda blastnr和uniprot是比较通用的数据库:
ftp://ftp.ncbi.nlm.nih.gov/blast/db/
https://www.uniprot.org/downloads
1)nr是ncbi收集的目前所有微生物的蛋白序列,是用来计算氨基酸一般情况下的频率的,160G
2)uniprot90根据相似性做了一个去冗余,所以比nr要小很多,56G
# 以uniprot90为例
wget ftp://ftp.uniprot.org/pub/databases/uniprot/uniref/uniref90/uniref90.fasta.gz # 下载
gzip -d uniref90.fasta.gz # 解压
makeblastdb -in uniref90.fasta -parse_seqids -hash_index -dbtype prot # 编译解析完成后的样子:
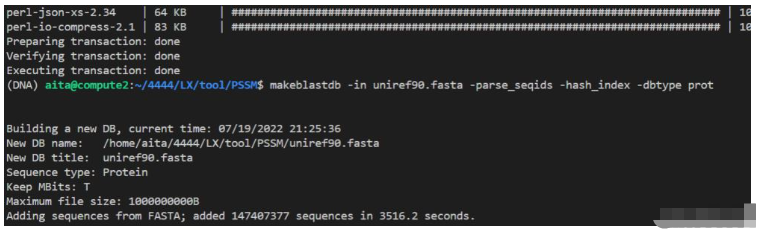
文件是这个样子:(只截取了一部分)
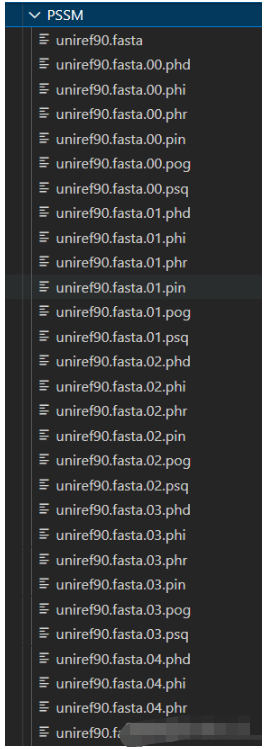
我的初始文件是:
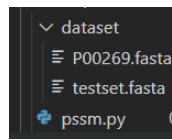
P00269.fasta是对单条蛋白质处理,里面的格式是:

testset.fasta是对蛋白质集合批处理,里面的格式是(也可以单独蛋白质存为.fasta文件,由于blast只能处理单条蛋白糊,把这个集合知识归总的意思,第一步还是要生成单条蛋白质的.fasta文件,所以这个文件看个人意愿):
import os
os.system('psiblast -query dataset/P00269.fasta -db /PSSM/uniref90.fasta -num_iterations 3 -out_ascii_pssm /dataset/P00269.pssm')##这个蛋白质好慢呀import os
file_name='/dataset/testset.fasta'
Protein_id=[]
with open(file_name,'r') as fp:
i=0
for line in fp:
if i%2==0:
# Protein_id.append(line[1:-1])
id=line[0:-1]
p=line[1:-1]
with open ('/dataset/'+str(p)+'.fasta','a') as protein:
protein.write(id)
# protein.write()
if i%2==1:
seq=line[0:-1]
with open ('/dataset/'+str(p)+'.fasta','a') as protein:
protein.write('\n')
protein.write(seq)
i=i+1
os.system('psiblast -query '+'/dataset/'+str(p)+'.fasta -db /PSSM/uniref90.fasta -num_iterations 3 -out_ascii_pssm /dataset/'+str(p)+'.pssm')##PSSM真是太慢了,下面是只生成一个后的截图
以上就是“怎么使用python服务器批处理得到PSSM矩阵”这篇文章的所有内容,感谢各位的阅读!相信大家阅读完这篇文章都有很大的收获,小编每天都会为大家更新不同的知识,如果还想学习更多的知识,请关注亿速云行业资讯频道。
亿速云「云服务器」,即开即用、新一代英特尔至强铂金CPU、三副本存储NVMe SSD云盘,价格低至29元/月。点击查看>>
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。



 计算
计算 安全
安全 数据库
数据库 网络和加速
网络和加速 企业服务
企业服务



