本篇文章给大家分享的是有关怎样使用R语言利用vcf格式文件计算核苷酸多样性,小编觉得挺实用的,因此分享给大家学习,希望大家阅读完这篇文章后可以有所收获,话不多说,跟着小编一起来看看吧。
bcftools view snp.vcf.gz scaffold_1 > popgenome-vcf/scaffold_1bcftools view snp.vcf.gz scaffold_2 > popgenome-vcf/scaffold_2
如果当前目录下只有vcf格式文件,会遇到报错Failed to open .vcf.gz: could not load index,可以参考 https://www.cnblogs.com/chenwenyan/p/11945445.html
tabix -p vcf snp.vcf.gz如果当前目录下没有popgenome-vcf这个目录,还需要新建目录
mkdir popgenome-vcf今天参考的文章里写道 In theory, the r PopGenome can read VCF files directly, using the readVCF function. However, because our samples are haploid, we need to use a different function, r readData, which requires a folder with a separate VCF for each scaffold. 这个是为什么呢?
#install.packages("PopGenome")library(PopGenome)getwd()setwd("VCF/")snp<-readData("popgenome-vcf",format = "VCF")get.sum.data(snp)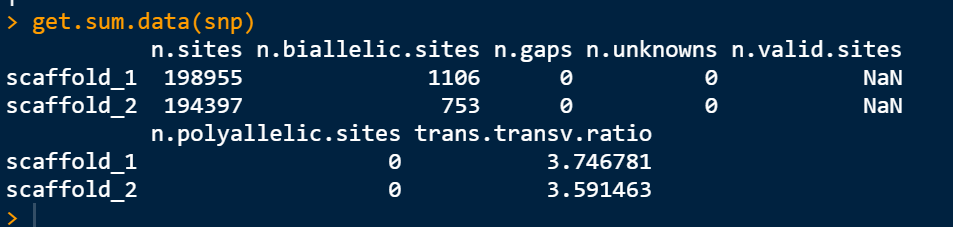
这里可以直接统计 转换和颠换的比例
pops<-get.individuals(snp)[[1]]pop1<-pops[grep("B\\.bam",pops)]pop2<-pops[grep("b\\.bam",pops)]pop1pop2snp<-set.populations(snp,list(pop1,pop2))snp@populationssnp<-F_ST.stats(snp)get.F_ST(snp)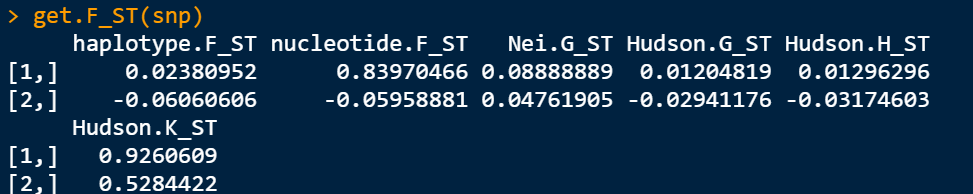
这里的指标都是什么意思呢?
get.diversity(snp)[[1]] 这里的指标也看不懂是什么意思呀
这里的指标也看不懂是什么意思呀
win_snp<-sliding.window.transform(snp, width = 10000, jump = 2000,type = 2)win_snp<-F_ST.stats(win_snp)win_snp@nucleotide.F_STwin_snp@nuc.diversity.withinlibrary(ggplot2)win_fst <- data.frame(x=1:dim(win_snp@nucleotide.F_ST)[1], y=win_snp@nucleotide.F_ST[,1])head(win_fst)p1<-ggplot(win_fst,aes(x=x,y=y))+ geom_point()+ geom_line()+ theme_bw()+ theme(panel.grid = element_blank())+ scale_x_continuous(breaks = win_fst$x, labels = win_fst$x)+ labs(x=NULL,y=NULL,title = "FST")ggsave("FST.pdf",p1,width = 15,height = 4)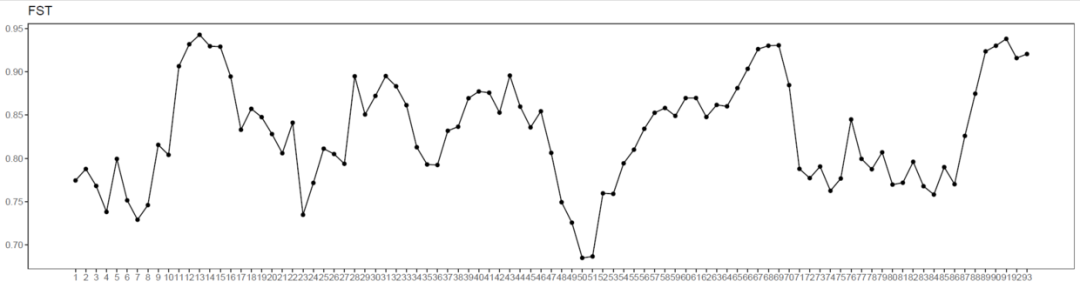
bb_div <- win_snp@nuc.diversity.within[,1] # diversity among B (bb = "big B")lb_div <- win_snp@nuc.diversity.within[,2] # diversity among B (lb = "little b")bb_divdf1<-data.frame(x=1:length(bb_div),y=bb_div)df2<-data.frame(x=1:length(lb_div),y=lb_div)p2<-ggplot()+ geom_line(data=df1,aes(x=x,y=y),color="red")+ geom_point(data=df1,aes(x=x,y=y),size=2,color="red")+ geom_line(data=df2,aes(x=x,y=y),color="blue")+ geom_point(data=df2,aes(x=x,y=y),size=2,color="blue")+ theme_bw()+labs(x=NULL,y=NULL)ggsave("diversity.pdf",p2,width = 15,height = 4)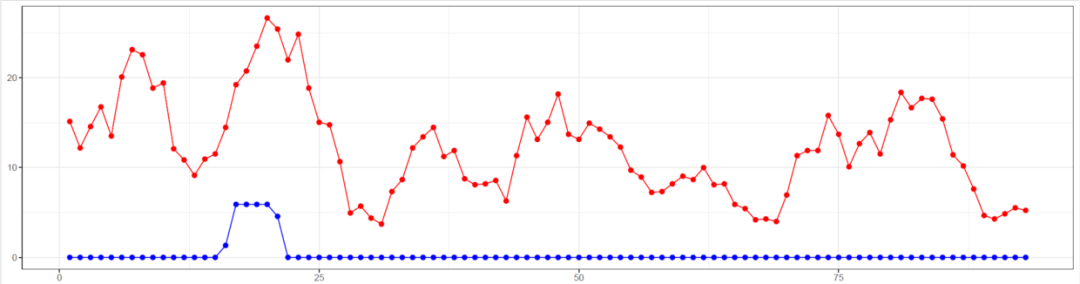
以上就是怎样使用R语言利用vcf格式文件计算核苷酸多样性,小编相信有部分知识点可能是我们日常工作会见到或用到的。希望你能通过这篇文章学到更多知识。更多详情敬请关注亿速云行业资讯频道。
亿速云「云服务器」,即开即用、新一代英特尔至强铂金CPU、三副本存储NVMe SSD云盘,价格低至29元/月。点击查看>>
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。
原文链接:https://my.oschina.net/u/4579431/blog/4949741



 计算
计算 安全
安全 数据库
数据库 网络和加速
网络和加速 企业服务
企业服务



