本篇文章为大家展示了如何理解基因组数据分析软件SpeedSeq,内容简明扼要并且容易理解,绝对能使你眼前一亮,通过这篇文章的详细介绍希望你能有所收获。
SpeedSeq是一款开源的基因组数据变异分析软件,主要功能如下
alignments, 序列比对
variant detection, 变异检测
functional annotation, 突变位点的功能注释
该软件最大的特点就是快速,对于50X的人类全基因组数据, 原始的fastq到vcf文件只需要13小时左右,对应的文章发表在nature methods上,链接如下
http://ucgd.genetics.utah.edu/wp-content/uploads/2015/08/nmeth.3505.pdf
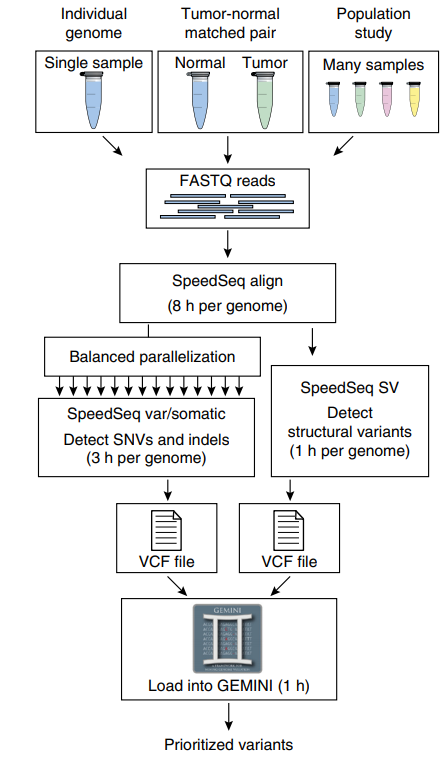
该软件是一个完整的pipeline, 集成了多款软件,可以用于检测以下多种基因组变异
germline and somatic mutations, 通过freebayes软件来检测突变微位点
structural variants,通过lumpy-sv软件来检测结构变异
其流程图示意如下
源代码保存在github上,链接如下
https://github.com/hall-lab/speedseq
该软件按照功能,拆分成了以下5个子模块
该模块将双端测序的fastq数据比对到参考基因组上,然后进行markduplicate, sort, index等步骤, 和GATK流程中的数据预处理步骤一致,用法如下
speedseq align \
-R "@RG\tID:sample1\tSM:sample1\tLB:sample1" \
-t 10 \
-o sample1 \
hg19.fa \
sample1_R1.fastq.gz \
sample1_R2.fastq.gz使用bwa软件比对参考基因组,然后使用samblaster进行markduplicate, sambamba软件进行bam文件的sort。
该模块用于检测生殖变异,输入为align模块产生的bam文件,用法如下
speedseq var \
-t 10 \
hg19.fa \
sample1.bam使用freebayes软件来检测生殖变异,输出文件为VCF文件。
该模块用于检测体细胞突变,输入为align模块产生的bam文件,用法如下
speedseq somatic \
-t 10 \
-o tumor \
hg19.fa \
normal.bam\
tumor.bam使用freebayes软件来检测体细胞突变,需要配对的肿瘤和正常样本,输出文件为VCF文件。
该模块用于检测结构变异,用法如下
speedseq sv \
-o sample \
-B sample.bam \
-D sample.discordants.bam \
-S sample.splitters.bam \
-R hg19.fa \
-o sample \
-t 10使用lumpy-sv软件来检测结构变异,输出文件为VCF文件。
该模块从bam文件中提取双端的fastq序列,再进行和align模块相同的处理,用法如下
speedseq realign \
-t 10 \
-o sample \
hg19.fa \
sample.ba要求bam文件必须包含read group信息,输出文件和align模块相同。对于全基因组数据的分析,使用speedseq可以大大加快处理速度。
上述内容就是如何理解基因组数据分析软件SpeedSeq,你们学到知识或技能了吗?如果还想学到更多技能或者丰富自己的知识储备,欢迎关注亿速云行业资讯频道。
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。





