这篇文章主要介绍“怎么使用CNVnator进行CNV检测”,在日常操作中,相信很多人在怎么使用CNVnator进行CNV检测问题上存在疑惑,小编查阅了各式资料,整理出简单好用的操作方法,希望对大家解答”怎么使用CNVnator进行CNV检测”的疑惑有所帮助!接下来,请跟着小编一起来学习吧!
CNVnator是一款CNV检测软件,基于Read-Depth的分析策略,通过对全基因组测序数据进行分析来预测CNV, 源代码保存在github上,网址如下
https://github.com/abyzovlab/CNVnator
这个软件的安装比较复杂,我这里直接使用别人装好的docker镜像进行处理,这也是docker的方便之处,直接从源中下载别人已经装好的cnvnator的镜像,代码如下
docker pull diploid/cnvnator
具体的分析步骤如下
CNVnator中依赖ROOT这个软件包,这个软件包是专门针对大数据的处理进行开发的,提供了统计分析,可视化,数据存储等一系列功能,基于这个体系可以极大的加快运行速度,所以将原始的信息都存在root文件中,便于分析。
这一步将比对的信息存储到后缀为root的文件中,代码如下
cnvnator -root file.root -tree chr1.bam -chrom 1
-tree参数指定输入的bam文件的名称,-root文件指定输出的root文件的名称,-chrom指定需要分析的染色体,默认情况下分析所有的染色体,这里我是测试,只分析了1号染色体。
这一步是按照固定大小的窗口对基因组进行划分,统计每个窗口内的read depth, 代码如下
cnvnator -root file.root -his 1000 -chrom 1 -d genome_fa_dir-his参数指定窗口的大小,单位为bp, -d参数指定参考基因组fasta文件所在的目录。
这一步是必须的,代码如下
cnvnator -root file.root -stat 1000-stat指定窗口的大小,和第二步的-his参数的取值相同。
这一步进行segmentation,代码如下
cnvnator -root file.root -partition 1000-partition指定窗口的大小,和第二步的-his参数的取值相同。
这一步分析每个segment区域的CNV, 代码如下
cnvnator -root file.root -call 1000 > cnv.call.txt-call指定窗口的大小,和第二步的-his参数的取值相同, 输出结果默认打印到屏幕上,这里重定向到文件中。
输出文件的内容如下
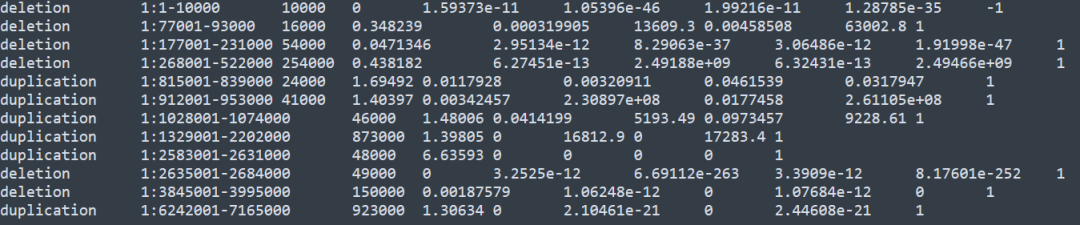
第一列为CNV的类型,包括了deletion和duplication两种类型,第二列为CNV的染色体区域,第三列为CNV的长度,第四列为归一化之后的read depth, 归一化到0-1的范围,第五列到第八列为不同的evalue值,最后一类为q0, 后五列对应的解释如下

对于原始的cnv call的输出,还可以通过软件自带的脚本转换为VCF格式,代码如下
cnvnator2VCF.pl cnv.call.txt >cnv.vcfCNVnator的功能强大,运行速度快,更多用法请参考官方文档。
到此,关于“怎么使用CNVnator进行CNV检测”的学习就结束了,希望能够解决大家的疑惑。理论与实践的搭配能更好的帮助大家学习,快去试试吧!若想继续学习更多相关知识,请继续关注亿速云网站,小编会继续努力为大家带来更多实用的文章!
亿速云「云服务器」,即开即用、新一代英特尔至强铂金CPU、三副本存储NVMe SSD云盘,价格低至29元/月。点击查看>>
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。
原文链接:https://my.oschina.net/u/4580290/blog/4570298



 计算
计算 安全
安全 数据库
数据库 网络和加速
网络和加速 企业服务
企业服务



