本篇内容介绍了“如何使用conifer进行WES的CNV分析”的有关知识,在实际案例的操作过程中,不少人都会遇到这样的困境,接下来就让小编带领大家学习一下如何处理这些情况吧!希望大家仔细阅读,能够学有所成!
和xhmm类似,conifer也是一款利用WES的数据来检测CNV的软件。不同的是,xhmm利用PCA算法达到降噪的目的,而conifer则通过SVD奇异值分解的算法来降噪,对应的文章链接如下
https://genome.cshlp.org/content/early/2012/05/14/gr.138115.112.full.pdf
官网如下
http://conifer.sourceforge.net/index.html
该软件的处理步骤示意如下
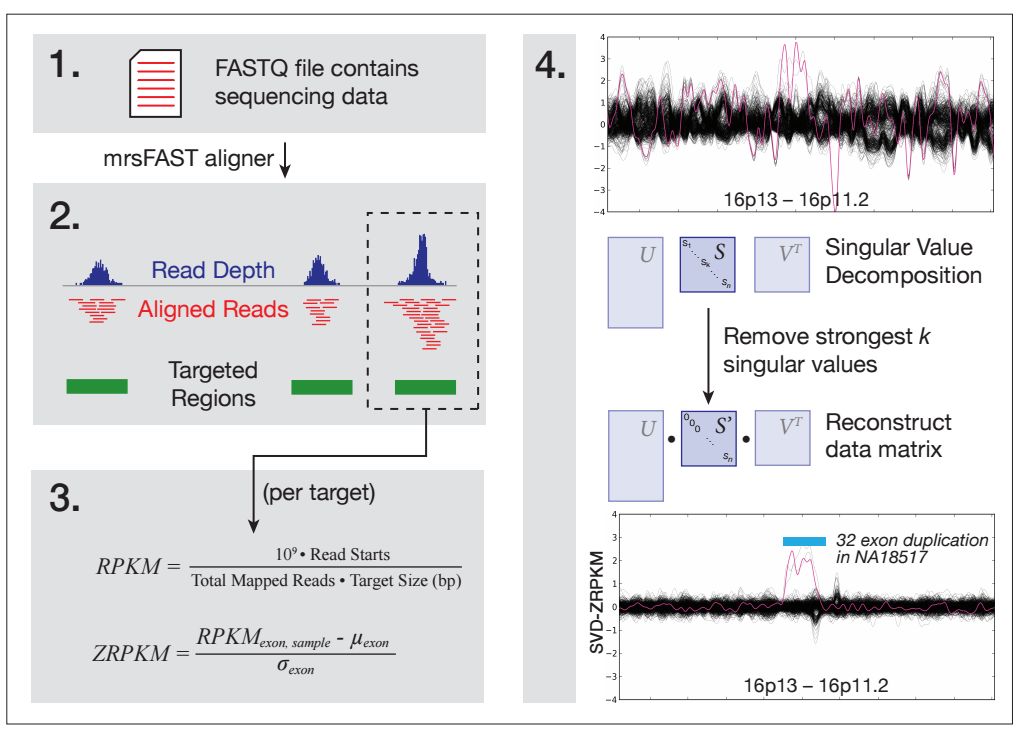
首先比对参考基因组,得到目标区域的测序深度,这里做了一个特殊的处理,借鉴了RNA_seq中定量的算法,计算了每个目标区域的RPKM值, 得到了所有样本每个目标区域RPKM值的矩阵,然后对矩阵进行标准化,计算z-score值,转换后的值称之为ZRPKM。
对于ZRPKM矩阵,采用SVD进行分解,认为奇异值大的子矩阵为系统噪声,去除奇异值大的子矩阵之后重新构建SVD-ZRPKM矩阵, 然后通过threshold calling算法预测CNV区域,图示如下
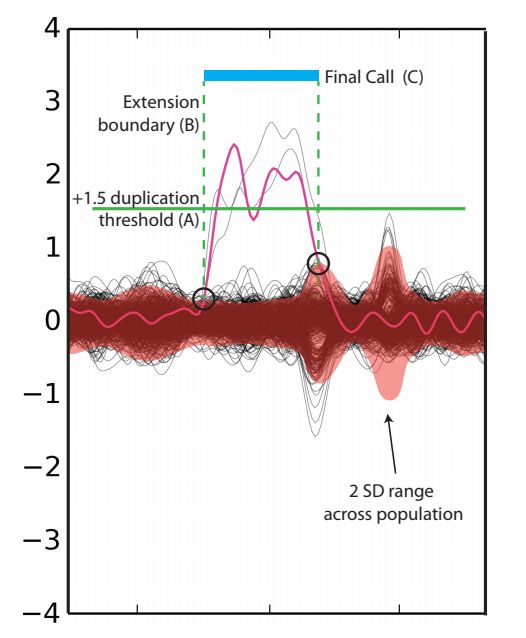
绿色线条表示阈值,大于1.5的认为是duplication, 小于1.5的认为是deletion。具体的步骤如下
首先基于芯片的捕获区域,创建目的区域对应的文件,内容如下
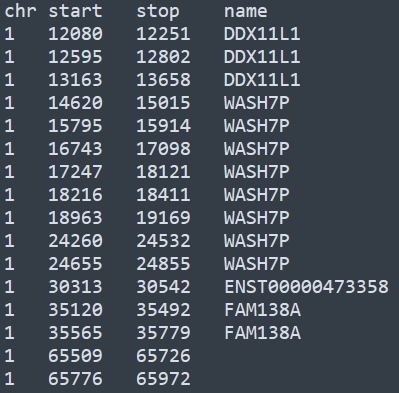
每一行代表一个捕获的目标区域,前三列对应目标区域的染色体位置,第四列为对应的基因名称,如果没有则为空。不同的捕获芯片对应的目的区域不同,自己写脚本整理成这种格式即可,下文中该文件称之为probes.txt。
对于每个样本,计算目标区域的RPKM值,用法如下
python conifer.py rpkm \--probes probes.txt \ --input sample1.bam \ --output RPKM/sample1.rpkm.txt输入文件为比对产生的bam文件,生物学重复的输出文件保存在同一个目录下。
读取所有样本的rpkm值,进行SVD奇异值分解,构建SVD-ZRPKM矩阵,用法如下
python conifer.py analyze \--probes probes.txt \--rpkm_dir ./RPKM/ \--output analysis.hdf5 \--svd 6 \--write_svals singular_values.txt \--plot_scree screeplot.png \--write_sd sd_values.txt输出文件为hdf5格式的文件,--svd参数指定需要去除的奇异值最大的topN个成分,这个值是需要动态调整的。
进行cnv calling, 用法如下
python conifer.py call \--input analysis.hdf5 \----threshold 1.5 \--output calls.txt输出文件内容示意如下

对感兴趣的CNV区域进行可视化,用法如下
python conifer.py plot \--input analysis.hdf5 \--region chr1:878657-889417 \--output image.png \--sample sampleA.rpkm可视化结果示意如下
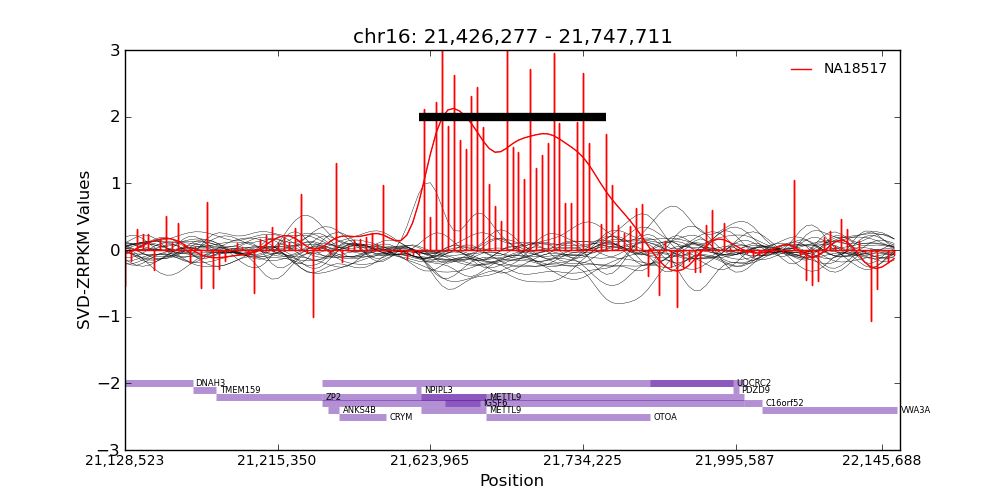
conifer用法简便, 适合检测1kb以上的CNV,软件要求CNV至少跨越3个exon区域,所以很短的CNV无法检测出来。
“如何使用conifer进行WES的CNV分析”的内容就介绍到这里了,感谢大家的阅读。如果想了解更多行业相关的知识可以关注亿速云网站,小编将为大家输出更多高质量的实用文章!
亿速云「云服务器」,即开即用、新一代英特尔至强铂金CPU、三副本存储NVMe SSD云盘,价格低至29元/月。点击查看>>
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。
原文链接:https://my.oschina.net/u/4580290/blog/4570270



 计算
计算 安全
安全 数据库
数据库 网络和加速
网络和加速 企业服务
企业服务



