本篇内容介绍了“tRNAscanSE怎么安装使用”的有关知识,在实际案例的操作过程中,不少人都会遇到这样的困境,接下来就让小编带领大家学习一下如何处理这些情况吧!希望大家仔细阅读,能够学有所成!
安装方法如下所示:
mkdir tRNAscantar -zxvf trnascan-se-2.0.5.tar.gzcd tRNAscan-SE-2.0./configure --prefix=(path)/tRNAscan #括号中省略了绝对路径makemake installecho 'export PATH=$PATH:(path)/tRNAscan/bin' >> ~/.bashrcsource ~/.bashrc
wget -c http://eddylab.org/software/infernal/infernal-1.1.2.tar.gzmkdir infernaltar -zxvf infernal-1.1.2.tar.gzcd infernal-1.1.2./configure --prefix=(path)/infernalmakemake installecho 'export PATH=$PATH:(path)/infernal/bin' >> ~/.bashrcsource ~/.bashrc
tRNAscan-SE -B -o tRNA.out -f rRNA.ss -m tRNA.stats genome.fasta-A 适合于古细菌;-B 适合于细菌;-G 适合于古细菌,细菌和真核生物的混合序列;-O 适合于线粒体和叶绿体。不设置以上选项,则默认适合于真核生物;-C 仅使用Cove 进行tRNA 分析。虽然从一定程度上提高了准确性,但是运行速度很慢;-o 结果文件名-f tRNA的二级结构结果文件名-m 统计结果文件名。
对细菌基因组序列进行预测,如下所示:
tRNAscan-SE -B -o twk.tRNA.out -f twk.tRNA.ss -m twk.tRNA.stats new.scaffolds.fasta
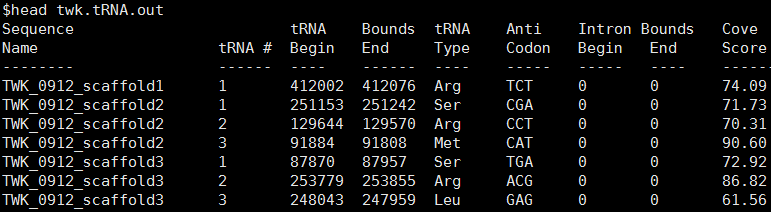
结果文件中out文件为不同Scaffolds上预测的tRNA位置及种类信息:
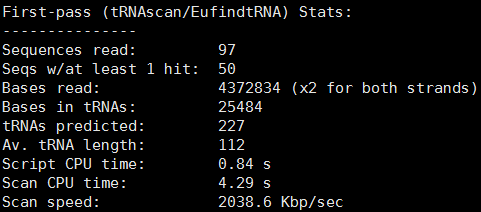
stats文件为预测到的tRNA统计信息,包括预测到的tRNA数、总碱基数等,如下所示:
ss文件为tRNA二级结构信息,如下所示:
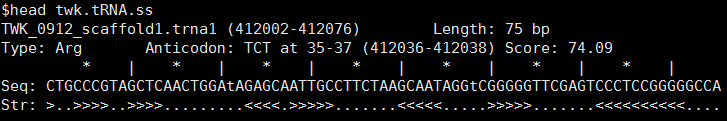
其中大于号或小于号表示互补配对区域,点号表示环形域或非互补配对区域。可以根据out文件与基因组序列提取出tRNA的序列文件与gff文件,如下所示:
perl 10_tRNAscan_parser.pl twk.tRNA.out new.scaffolds.fasta TWK
结果如下所示:

“tRNAscanSE怎么安装使用”的内容就介绍到这里了,感谢大家的阅读。如果想了解更多行业相关的知识可以关注亿速云网站,小编将为大家输出更多高质量的实用文章!
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。





