VCF转换PLINK格式的3种方法是什么,相信很多没有经验的人对此束手无策,为此本文总结了问题出现的原因和解决方法,通过这篇文章希望你能解决这个问题。
plink是目前使用的最为广泛的关联分析软件,其定义的ped/map文件系统,及其对应的二进制bed/bim/fam已经成为关联分析的标准文件格式。在进行关联分析之前,我们首先要做的就是将其他格式的文件转换为plink对应的文件格式。
VCF格式作为存储分型结果的一种标准格式,在实际分析中也广泛应用。本文总结了将vcf文件转换为plink对应文件格式的3种方式,详细展示如下
在gatk3中,提供了一个名为VariantsToBinaryPed的功能,可以将VCF格式转换为plink对应的二进制bed文件,基本用法如下
java -jar GenomeAnalysisTK.jar \-T VariantsToBinaryPed \-R reference.fasta \-V input.vcf \-m input.fam \-bed output.bed \-bim output.bim \-fam output.fam要求三个输入文件,-R参数指定参考基因组的fasta文件,-V参数指定VCF文件,-m参数称之为metadata, 保存了样本对应的家系信息,支持两种文件格式,第一种示意如下
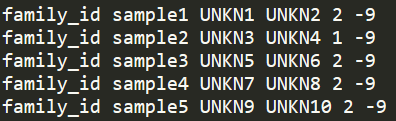
对应ped文件的前六列内容,如果样本双亲信息不明确,则用unknown表示,简写成UNKN, 表型信息没有的话用-9表示。第二种示意如下
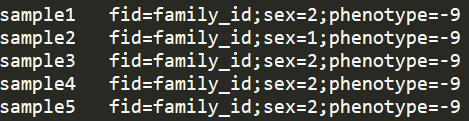
由于VCF文件中只保存了样本的分型结果,所以家系信息通过一个额外的文件来补充。更多详情请参考以下文档
https://software.broadinstitute.org/gatk/documentation/tooldocs/3.8-0/org_broadinstitute_gatk_tools_walkers_variantutils_VariantsToBinaryPed.php
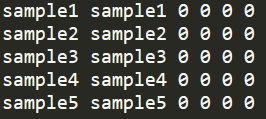
vcftools是操作vcf文件的一款常用工具,支持将vcf文件转换成plink对应的ped/map格式,基本用法如下
vcftools --vcf input.vcf --plink --out output没有额外的家系信息,在输出结果中,family id和sample id是重复的,其他列全部为0,示意如下
plink1.9版本支持直接读取vcf/gen等多种文件格式,所以使用该版本时其实不需要专门进行格式转换,软件默认会将不同的格式转换为二进制bed文件格式。这里只是展示下其格式转换的用法,基本用法如下
plink --vcf input.vcf --recode --out output --double-id默认转换为二进制的bed格式,对于分析更加适用,这里添加--recode参数将输出结果调整为ped格式,可以更好的查看其转换的规则。
plink默认用下划线对样本名进行分隔,分隔的两个字段分别作为ped文件中的family id和sample id, 如果vcf中的样本名含有多个下划线,无法正确进行划分,软件会报错,此时可以修改--id-delim参数,该参数设定了分隔符,默认是下划线,可以设置成其他字符,以达到正确区分的目的。
除此之外,还有一种解决方案,通过参数指定family_id的设定方式,有两种参数,第一种如上述示例中的--double_id, 将family id和sample id保持相同,输出的ped文件的前六列示意如下
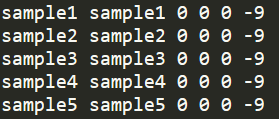
第二种参数用法如下所示
plink --vcf input.vcf --recode --out output --const-fid family_id通过--const-fid将family id设置成一个常量,默认值是0,上述示例输出的ped文件的前六列示意如下
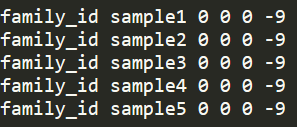
双亲,性别默认用0填充,表型默认用-9填充。
看完上述内容,你们掌握VCF转换PLINK格式的3种方法是什么的方法了吗?如果还想学到更多技能或想了解更多相关内容,欢迎关注亿速云行业资讯频道,感谢各位的阅读!
亿速云「云服务器」,即开即用、新一代英特尔至强铂金CPU、三副本存储NVMe SSD云盘,价格低至29元/月。点击查看>>
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。
原文链接:https://my.oschina.net/u/4580290/blog/4357154



 计算
计算 安全
安全 数据库
数据库 网络和加速
网络和加速 企业服务
企业服务



