本篇内容介绍了“如何利用bedtools预测chip_seq数据的靶基因”的有关知识,在实际案例的操作过程中,不少人都会遇到这样的困境,接下来就让小编带领大家学习一下如何处理这些情况吧!希望大家仔细阅读,能够学有所成!
通常在分析peak区域对应的靶基因时,会选取转录起始位点TSS上下游一定长度的区域作为候选的靶基因范围,本文介绍下如何利用bedtools来对peak与TSS区域的overlap情况进行分析,从而得到靶基因,可以分为以下几步
以hg38为例,通过UCSC的FTP服务可以得到物种对应的refFlat文件,链接如下
http://hgdownload.soe.ucsc.edu/goldenPath/hg38/database/

refFLat和refGene这两个文件记录的信息相同,refFlat文件列数更少,这里我们选择下载refFlat.txt.gz, 该文件的内容如下所示
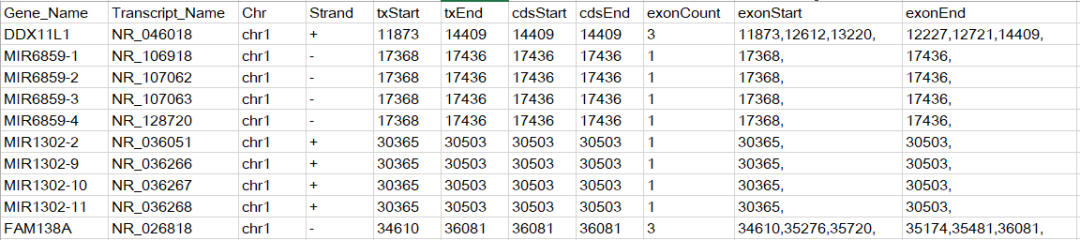
在原始文件中是没有第一行的标题的,我手动添加的标题是为了方便描述每列的含义,从该文件中可以得到TSS位点信息。
bedtools要求输入的文件格式为bed, gff, vcf等,这里我们需要把上述下载的原始文件转换为bed格式,用法如下
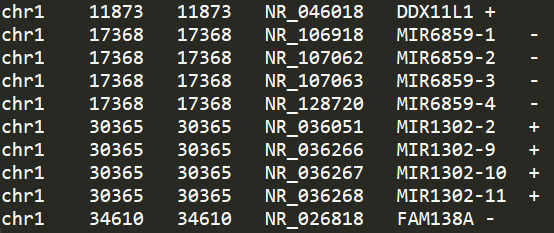
awk '{print $3"\t"$5"\t"$5"\t"$2"\t"$1"\t"$4}' > hg38.tss.bed内容如下所示
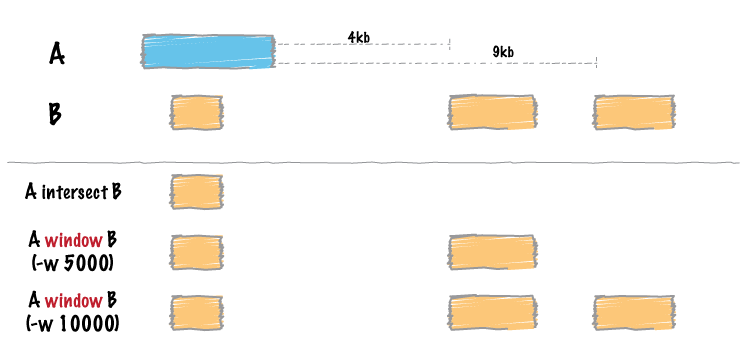
bedtools windows和intersect的功能类似,都是用于求两个区间A和B的交集,只不过window会在A区间的上下游加上一个可以自定义的长度之后,再与B区间求交集,原理示意如下
以TSS上下游5kb为例,用法如下
bedtools window -a hg39.tss.bed -b peak.bed -w 5000 -sm > overlap.txt
通过window这个命令,可以灵活的定义TSS上下游的区间,快速得到peak对应的靶基因。
“如何利用bedtools预测chip_seq数据的靶基因”的内容就介绍到这里了,感谢大家的阅读。如果想了解更多行业相关的知识可以关注亿速云网站,小编将为大家输出更多高质量的实用文章!
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。





