本篇内容主要讲解“r语言怎么实现LM模型+数值+因子协变量”,感兴趣的朋友不妨来看看。本文介绍的方法操作简单快捷,实用性强。下面就让小编来带大家学习“r语言怎么实现LM模型+数值+因子协变量”吧!
第一列为FID 第二列为ID 第三列以后为协变量(注意,只能是数字,不能是字符!)
这里协变量文件为:
[dengfei@ny 03_linear_cov]$ head cov.txt 1061 1061 F 31062 1062 M 31063 1063 F 31064 1064 F 31065 1065 F 31066 1066 F 31067 1067 F 31068 1068 M 31069 1069 M 31070 1070 M 3sed 's/F/1/g' cov.txt >cov2.txtsed -i 's/M/2/g' cov2.txt 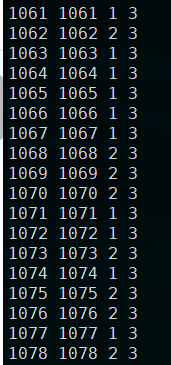
plink --file b --covar cov2.txt --write-covar --dummy-coding结果生成:
plink.cov
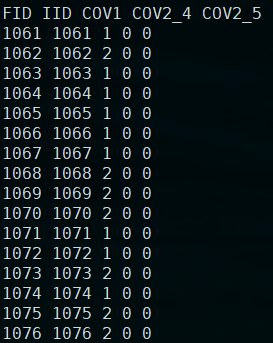
「注意:」这里的性别虽然是因子,但是其只有两个水平,也可以将作为连续的变量,计算方法是一样的。如果是三个水平的因子,就不能直接转化为变量了。
「代码:」
plink --file b --pheno phe.txt --allow-no-sex --linear --covar plink.cov --out re --hide-covar「日志:」
PLINK v1.90b5.3 64-bit (21 Feb 2018) www.cog-genomics.org/plink/1.9/(C) 2005-2018 Shaun Purcell, Christopher Chang GNU General Public License v3Logging to re.log.Options in effect: --allow-no-sex --covar plink.cov --file b --hide-covar --linear --out re --pheno phe.txtNote: --hide-covar flag deprecated. Use e.g. '--linear hide-covar'.515199 MB RAM detected; reserving 257599 MB for main workspace..ped scan complete (for binary autoconversion).Performing single-pass .bed write (10000 variants, 1500 people).--file: re-temporary.bed + re-temporary.bim + re-temporary.fam written.10000 variants loaded from .bim file.1500 people (0 males, 0 females, 1500 ambiguous) loaded from .fam.Ambiguous sex IDs written to re.nosex .1500 phenotype values present after --pheno.Using 1 thread (no multithreaded calculations invoked).--covar: 3 covariates loaded.Before main variant filters, 1500 founders and 0 nonfounders present.Calculating allele frequencies... done.10000 variants and 1500 people pass filters and QC.Phenotype data is quantitative.Writing linear model association results to re.assoc.linear ... done.「结果文件:」re.assoc.linear
「结果预览:」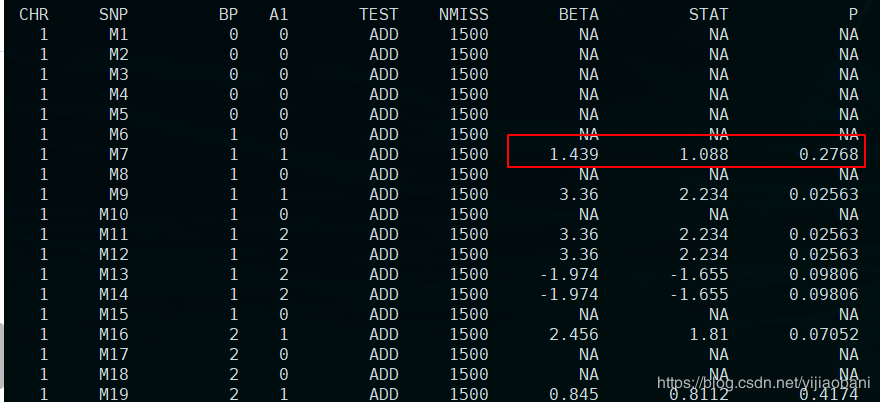
library(data.table)geno = fread("c.raw")geno[1:10,1:10]phe = fread("phe.txt")cov = fread("cov.txt")plink = fread("plink.cov")dd = data.frame(phe = phe$V3,cov1 = plink$COV1,cov2 = plink$COV2_4,cov3=plink$COV2_5,geno[,7:20])head(dd)mod_M7 = lm(phe ~ cov1+cov2+cov3 + M7_1,data=dd);summary(mod_M7)「M7加上因子协变量结果:」
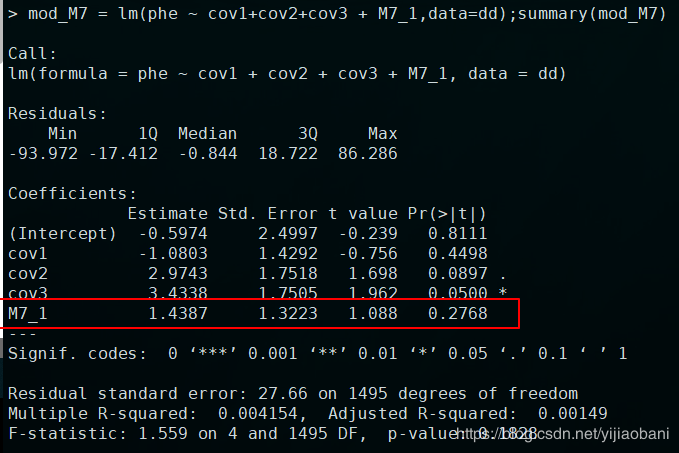
「这里,我们可以测试一下:」将性别由数字,变为因子,可以发现结果是一样的:
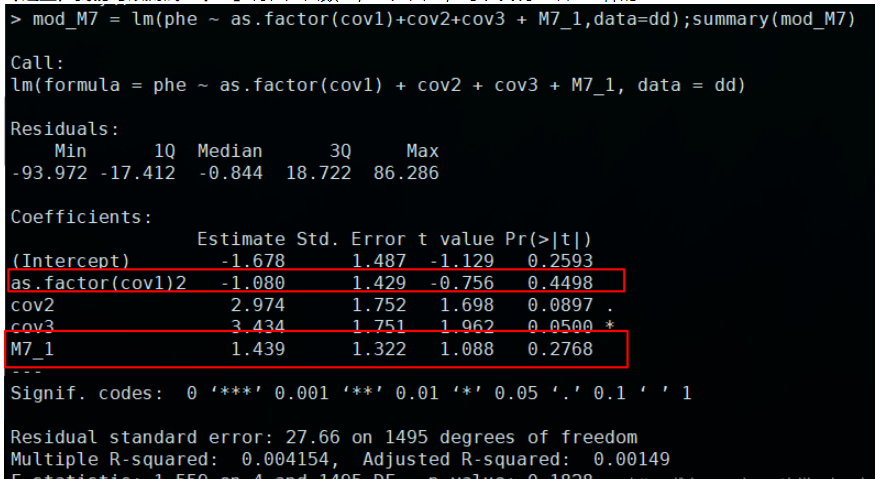
到此,相信大家对“r语言怎么实现LM模型+数值+因子协变量”有了更深的了解,不妨来实际操作一番吧!这里是亿速云网站,更多相关内容可以进入相关频道进行查询,关注我们,继续学习!
亿速云「云服务器」,即开即用、新一代英特尔至强铂金CPU、三副本存储NVMe SSD云盘,价格低至29元/月。点击查看>>
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。
原文链接:https://my.oschina.net/u/4592498/blog/4451027



 计算
计算 安全
安全 数据库
数据库 网络和加速
网络和加速 企业服务
企业服务



