GSEA软件的使用方法是什么,针对这个问题,这篇文章详细介绍了相对应的分析和解答,希望可以帮助更多想解决这个问题的小伙伴找到更简单易行的方法。
Gene Set Enrichment Analysis是一种富集算法,由Broad Institute研究所的科学家提出,算法核心示意如下
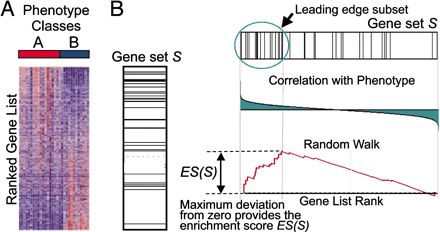
需要两个输入元素,一个就是排序好的基因列表,这里的排序的规则是展现两组间的差异,比如按照Foldchange的值进行排序,第二个就是基因的注释集合,然后运行KS检验计算Enrichment Score(ES),用置换检验评估ES的可靠性。
Broad Institute研究所的科学家同时还提供对应的分析软件GSEA,该软件是java语言开发的图形界面软件,简单易用,下载地址如下
http://software.broadinstitute.org/gsea/downloads.jsp
官网提供了多种下载方式,推荐直接下载jar文件,示意图如下

如下所示,运行GSEA分析,需要两个基本元素,第一个就是表达谱数据,可以是芯片数据,也可以是rna-seq的定量结果,第二个就是基因集数据库,官网对于human提供了MSigDB数据库,当然你也可以自己定义基因集。
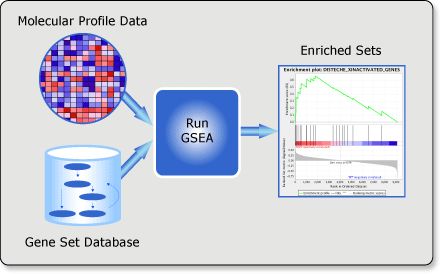
在实际操作时,第一步首先是导入数据,有以下4种数据需要导入,由于在windows平台操作,通过特定的后缀来识别文件格式
表达量文件,可以是芯片,也可以是rna-seq的定量结果,后缀为gct, 示意如下
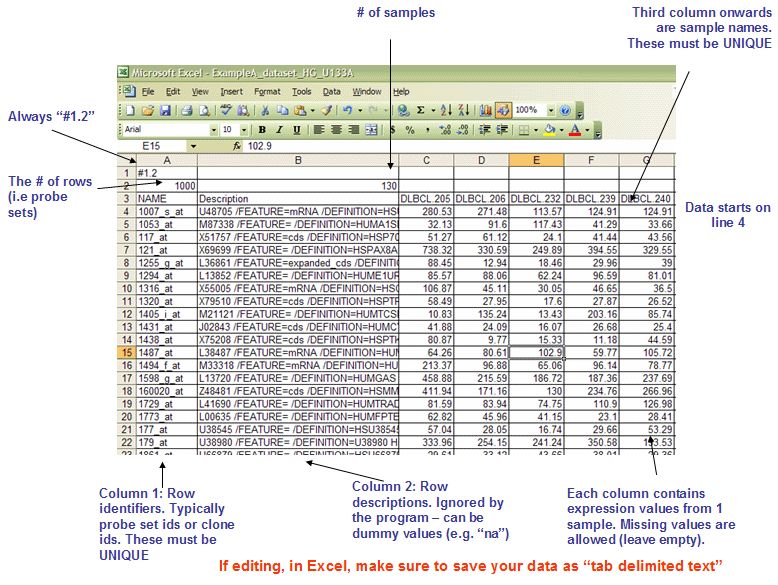
该文件是\t分隔的纯文本文件,第一行内容总是为#1.2, 表示版本,第二行表示表达量矩阵的维度,第一个值对应探针探针/基因个数,第二个数值代表样本个数,第三行是表达量矩阵的表头,前两列固定是NAME和Description, NAME是基因ID或者探针ID,必须保证唯一,Description表示描述信息,如果没有,可以用na填充,后面每列对应一个样本。
样本的分组文件,后缀为cls, 示意如下
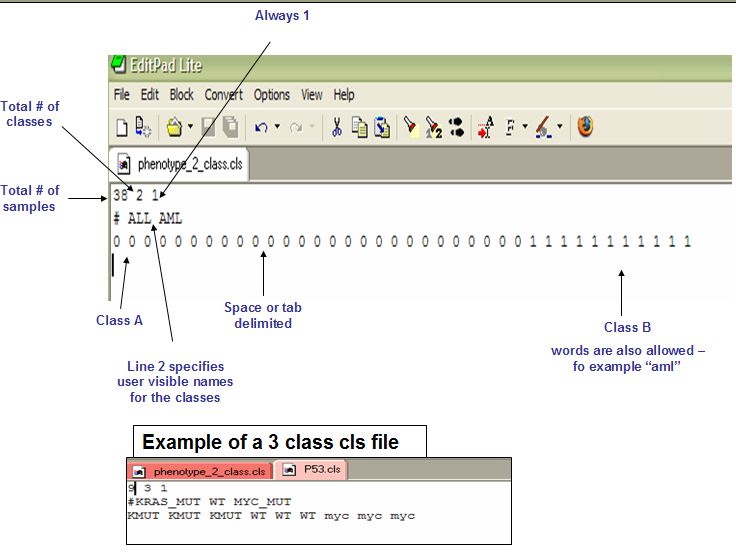
第一行为空格或者\t分隔的3个数值,第一个数值表示样本总数,第二个数值表示样本对应的分组数目,第三个数值总是1。
第二行以#开头,指定不同分组的名字;第三行的每个字段代表一个样本,顺序和表达量文件中的样本顺序一致,只不过将样本名用对应的分组名字表示。
基因集文件,有多种格式,常用的有gmt和gmx, gmt示意如下
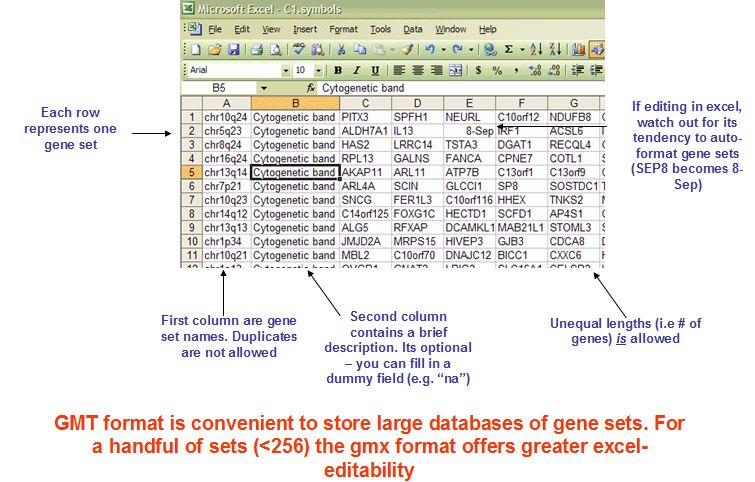
每一行代表一个基因集合,第一列为基因集合的名字,必须唯一,第二列为描述信息,如果没有就用na填充,后面的列为该集合下的基因,每列之间用\t分隔。gmt格式示意如下
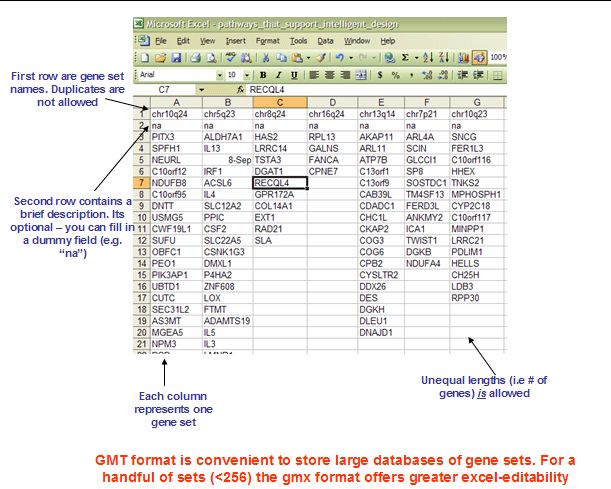
和gmt相反,gmt中每一列代表一个基因集合,第一行为基因集合的名字,必须唯一,第二行为描述信息,如果没有就用na填充,其他行为该集合下的基因。
当提供了芯片数据时,可以导入chip类型的文件,该文件保存的是探针和基因之间的对应关系,后缀为chip, 示意如下

第一列为探针ID, 表头为Probe_Set_ID,第二列为探针对应的基因,表头为Gene Symbol, 第三列为探针描述信息,没有就用na填充。
通过Load Data,首先将以上几种文件导入软件中,然后点击Run GSEA菜单,选择对应的各种文件

phenotype labels用于指定组间比较的顺序,明确哪一组作为control组。
上文中提到,GSEA需要两个输入元素,排序好的基因列表和基因集合,当导入表达量数据和分组信息后,GSEA会自动计算分组将的差异值,然后根据这个差异值对基因进行排序,支持的统计量有以下几种,其中
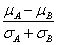
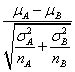
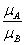
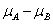
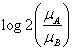
默认的算法为signal2noise, 可以在Basic fields中进行调整,这个参数可以在``示意如下

当所有参数都设置好之后,点击下方的Run按钮即可运行。
关于GSEA软件的使用方法是什么问题的解答就分享到这里了,希望以上内容可以对大家有一定的帮助,如果你还有很多疑惑没有解开,可以关注亿速云行业资讯频道了解更多相关知识。
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。





