本篇文章给大家分享的是有关miRNA定量原理是什么,小编觉得挺实用的,因此分享给大家学习,希望大家阅读完这篇文章后可以有所收获,话不多说,跟着小编一起来看看吧。
在转录组的数据分析中,定量和差异分析是基础分析内容,对于mRNA的定量,直接将reads比对参考基因组进行定量即可,但是对于miRNA数据而言,这样的操作方式就不合适了。
miRNA长度在18bp-36b之间,非常的短,如果用这么短的reads比对整个基因组,肯定会有多个比对上的位置,在定量时,无法有效区分reads到底来自于哪一个miRNA,而且gtf文件中只会记录miRNA基因的基因组位置,并不提供5’和3’端两个成熟的miRNA的基因组位置。
为了准确对miRNA进行定量,mirdeep软件的开发者提出了这样一种思路,将reads 比对miRNA的前体序列,这样可以有效判断reads到底属于哪一个miRNA;其次,考虑到一个miRNA前体可以产生两个成熟的miRNA,根据reads的比对位置来区分两个成熟的miRNA, 示意图如下
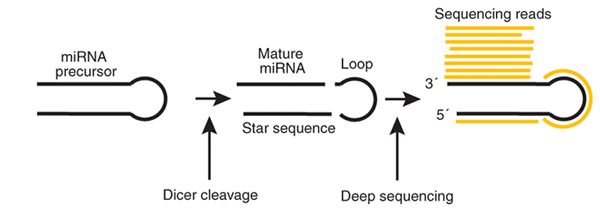
在最开始研究miRNA的时候,发现一个miRNA前体可以产生两个小的RNA, 将其中表达量高的一个称之为mature miRNA, 将表达量相对较低的称之为star miRNA, 在后来直接将两个RNA都称之为mature miRNA了,mirdeep软件中依然采用了star序列的命名方式,实际是都是miRNA的序列。
在实际操作时,通过bowtie1进行比对,bowtie1对于短序列的比对任务灵敏度更高,适用于miRNA这种短序列的比对工作,首先对miRNA前体构建索引,代码如下
bowtie-build precursor.converted miRNA_precursor
接下来,将成熟的miRNA比对到miRNA前体上,代码如下
bowtie -p $threads -f -v 0 -a --best --strata --norc miRNA_precursor mature.converted ${name1}_mapped.bwt最后,将miRNA测序的reads比对到miRNA前体上,代码如下
bowtie -p $threads -f -v $mismatches -a --best --strata --norc miRNA_precursor $name2.converted ${name2}_mapped.bwt根据两个比对产生的结果文件,就可以对miRNA进行定量,首先从成熟miRNA的比对结果中,确定成熟miRNA在miRNA前体上的位置, 比对结果示意如下
hsa-miR-550b-2-5p + hsa-mir-550b-1 15 ATGTGCCTGAGGGAGTAAGACA IIIIIIIIIIIIIIIIIIIIII 1 hsa-miR-550b-3p + hsa-mir-550b-1 57 TCTTACTCCCTCAGGCACTG IIIIIIIIIIIIIIIIIIII 1
对于hsa-mir-550b-1这个前体而言,有hsa-miR-550b-2-5p和hsa-miR-550b-3p两个成熟的miRNA,hsa-miR-550b-2-5p比对到了前体的15-36bp的位置,hsa-miR-550b-3p比对到前体的57-76bp的位置,这个位置是根据第4列的值加上第五列的序列长度得到的。
接下来从测序reads的比对结果,确定每个miRNA的表达量,比对结果示意如下
SW1_224845_x3 + hsa-mir-550b-1 17 GTGCCTGAGGGAGTAAGAGA IIIIIIIIIIIIIIIIIIII 4 18:C>G
SW1_287750_x2 + hsa-mir-550b-1 18 TGCCTGAGGGAGTAAGAGA IIIIIIIIIIIIIIIIIII 4 17:C>G
SW2_152540_x1 + hsa-mir-550b-1 17 GTGCCTGAGGGAGTAAGACT IIIIIIIIIIIIIIIIIIII 2 19:A>T
SW2_152541_x7 + hsa-mir-550b-1 17 GTGCCTGAGGGAGTAAGAGA IIIIIIIIIIIIIIIIIIII 4 18:C>G
SW2_199319_x1 + hsa-mir-550b-1 18 TGCCTGAGGGAGTAAGAA IIIIIIIIIIIIIIIIII 4 17:C>A
SW3_404761_x1 + hsa-mir-550b-1 17 GTGCCTGAGGGAGTAAGAGA IIIIIIIIIIIIIIIIIIII 4 18:C>G
SW3_516112_x1 + hsa-mir-550b-1 18 TGCCTGAGGGAGTAAGAGA IIIIIIIIIIIIIIIIIII 4 17:C>G
SW4_451132_x4 + hsa-mir-550b-1 17 GTGCCTGAGGGAGTAAGAGA IIIIIIIIIIIIIIIIIIII 4 18:C>G
SW4_589021_x1 + hsa-mir-550b-1 18 TGCCTGAGGGAGTAAGAGA IIIIIIIIIIIIIIIIIII 4 17:C>G我们可以看到,在实际测序的样本中,有这么多reads比对到了hsa-mir-550b-1这个前体;接下来的任务就是区分这些reads到底属于哪些成熟的miRNA。
从比对结果可以很明显看到,实际测序的reads在前体上的位置都是在17-36bp之间,对于这个区间而言,只能是hsa-miR-550b-2-5p这个成熟的miRNA。
为了加快比对过程,mirdeep对测序reads进行了处理,合并了冗余reads, 用序列标识符中x后面的数字代表在原始测序结果中该reads出现的次数,最终计数时,直接把x后面对应的数字相加就可以了。
mirdeep软件是miRNA分析中非常经典的一款软件,了解其定量原理有助于我们更好的理解miRNA数据分析过程。
以上就是miRNA定量原理是什么,小编相信有部分知识点可能是我们日常工作会见到或用到的。希望你能通过这篇文章学到更多知识。更多详情敬请关注亿速云行业资讯频道。
亿速云「云服务器」,即开即用、新一代英特尔至强铂金CPU、三副本存储NVMe SSD云盘,价格低至29元/月。点击查看>>
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。
原文链接:https://my.oschina.net/u/4580290/blog/4616016



 计算
计算 安全
安全 数据库
数据库 网络和加速
网络和加速 企业服务
企业服务



