小编给大家分享一下CPAT软件有什么用,希望大家阅读完这篇文章之后都有所收获,下面让我们一起去探讨吧!
随着高通量测序在lncRNA研究领域的应用, 越来越多的lncRNA被发现。对于转录组测序的数据而言,组装得到转录本之后,首先要做的就是区分蛋白编码和非蛋白编码的RNA。
目前针对这一问题,有多种解决方案,基本可以分为以下两类
alignment-based
alignment-free
第一种算法基于序列比对,可以较好的识别保守性较好的蛋白编码基因, 包括CPC,PhyloCSF等软件; 第二种算法不需要比对,而是通过coding和non-coding转录本的序列特征来进行区分,包括CNCI, CPAT, PLEK等。
lncRNA在物种间的保守性较差,另外部分lncRNA的染色体位置和蛋白编码基因存在重叠,通过序列比对的方式来区分容易造成误判。除此之外,基于序列比对的软件,其运行速度相对较慢,所以采用第二种算法的软件综合效果更好。
本文主要介绍CPAT的使用,网址如下
http://lilab.research.bcm.edu/cpat/
对于一个转录本而言,它是coding还是noncoding, 本质上是一个二分类问题,所以CPAT的开发者想到了通过逻辑回归来解决这个问题。该软件基于以下四个特征构建了逻辑回归模型来区分coding和noncoding
open reading frame size
open reading frame coverage
Fickett TESTCODE statistic
hexamer usage bias
前两个因素都是针对开放阅读框定义的,第一个因素是开放阅读框的大小,第二个因素是开放阅读框占转录本总长度的比例,第三个因素基于序列的碱基组成和密码子分布进行定义,第四个因素基于序列中六聚体的频率进行定义。
在论文中,针对以上4种特征,首先评估在coding和noncoding中的分布,图示如下
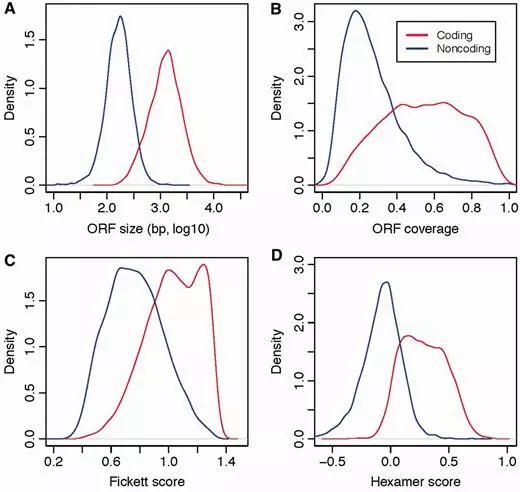
可以看到,coding和noncoding形成了两个不同的峰,说明这4种特征在coding和noncoding之间确实存在差异。
在论文中还通过ROC曲线评估了不同软件的性能,结果如下
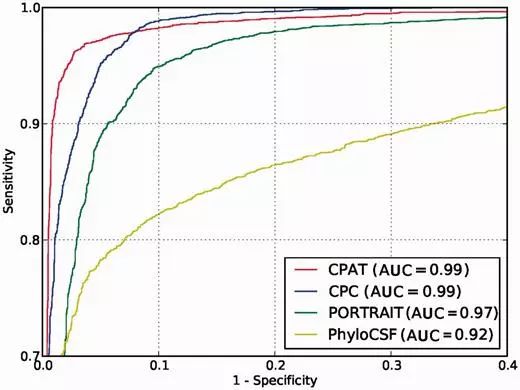
可以看到CPAT和CPC的效果是最好的。CPAT基于python编程语言开发,安装非常的简便,代码如下
pip install CPAT
该软件既可以在本地运行,也提供了在线版本。
在线版本的网址如下
http://lilab.research.bcm.edu/cpat/
可以直接输入fasta格式的序列,也可以输入bed格式的文件,此时需要指定对应的基因组版本,示意如下
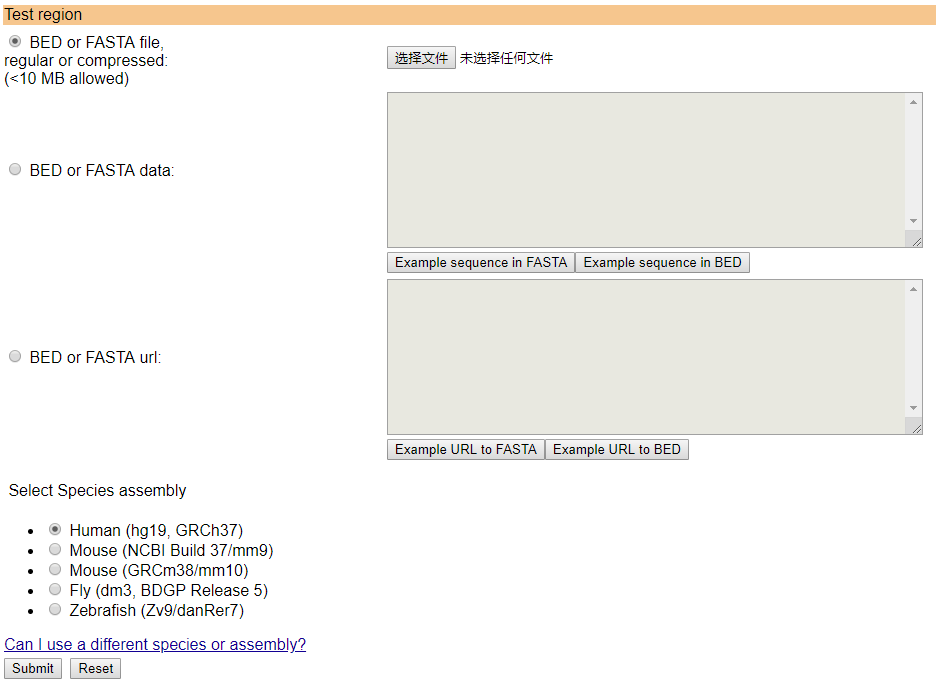
本地版本对应的也有两种用法,输入bed文件的用法如下
cpat.py -r /database/hg19.fa \
-g mRNA_hg19.bed \
-d dat/Human_logitModel.RData \
-x dat/Human_Hexamer.tsv \
-o output.txt输入fasta文件的用法如下
cpat.py -g transcript.fa \
-d dat/Human_logitModel.RData \
-x dat/Human_Hexamer.tsv \
-o output.txt-d和-x参数对应的文件为软件构建好的模型,位于软件的安装目录下。软件的输出结果如下所示

最后一列给出了转录本的蛋白编码信息,yes代表该转录本为protein-coding转录本,no代表该转录本为noncoding转录本。
看完了这篇文章,相信你对“CPAT软件有什么用”有了一定的了解,如果想了解更多相关知识,欢迎关注亿速云行业资讯频道,感谢各位的阅读!
亿速云「云服务器」,即开即用、新一代英特尔至强铂金CPU、三副本存储NVMe SSD云盘,价格低至29元/月。点击查看>>
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。
原文链接:https://my.oschina.net/u/4580290/blog/4602651



 计算
计算 安全
安全 数据库
数据库 网络和加速
网络和加速 企业服务
企业服务



