这篇文章主要讲解了“如何使用TCGAbiolinks分析TCGA中的表达谱数据”,文中的讲解内容简单清晰,易于学习与理解,下面请大家跟着小编的思路慢慢深入,一起来研究和学习“如何使用TCGAbiolinks分析TCGA中的表达谱数据”吧!
对于转录组数据而言,差异分析和富集分析是最核心的分析内容之一,通过TCGAbiolinks可以轻松实现TCGA表达谱数据的下载,差异分析,富集分析等功能,以乳腺癌的基因表达谱为例,分析过程如下
由于TCGA中乳腺癌的样本很多,所以只挑选了部分样本来测试,下载的过程如下
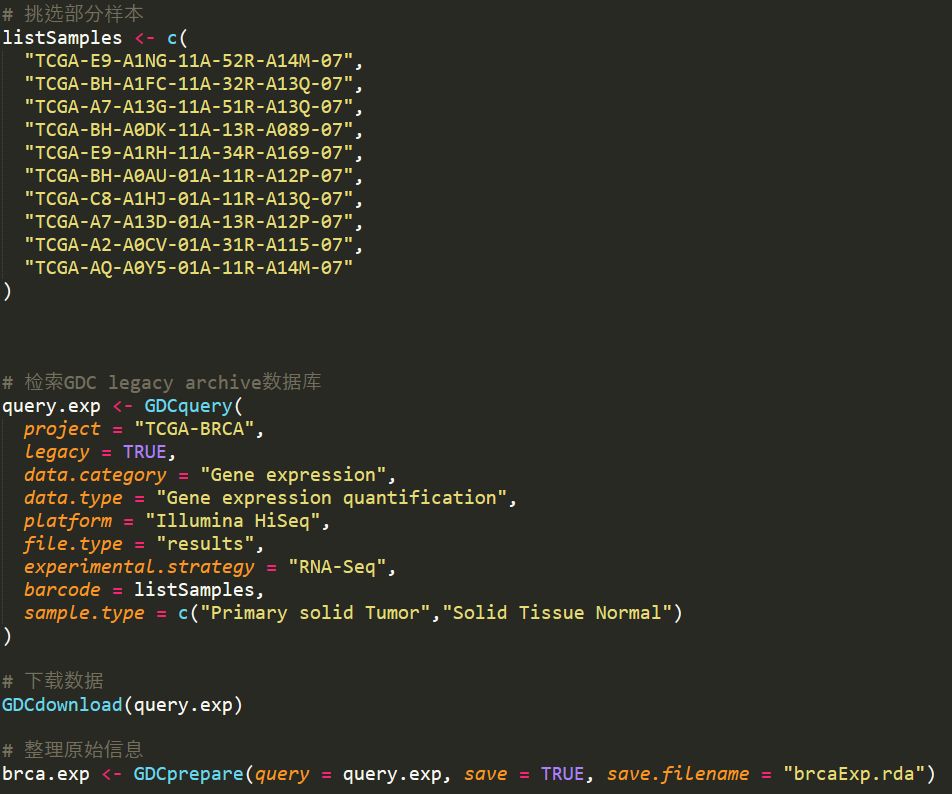
详细步骤如下
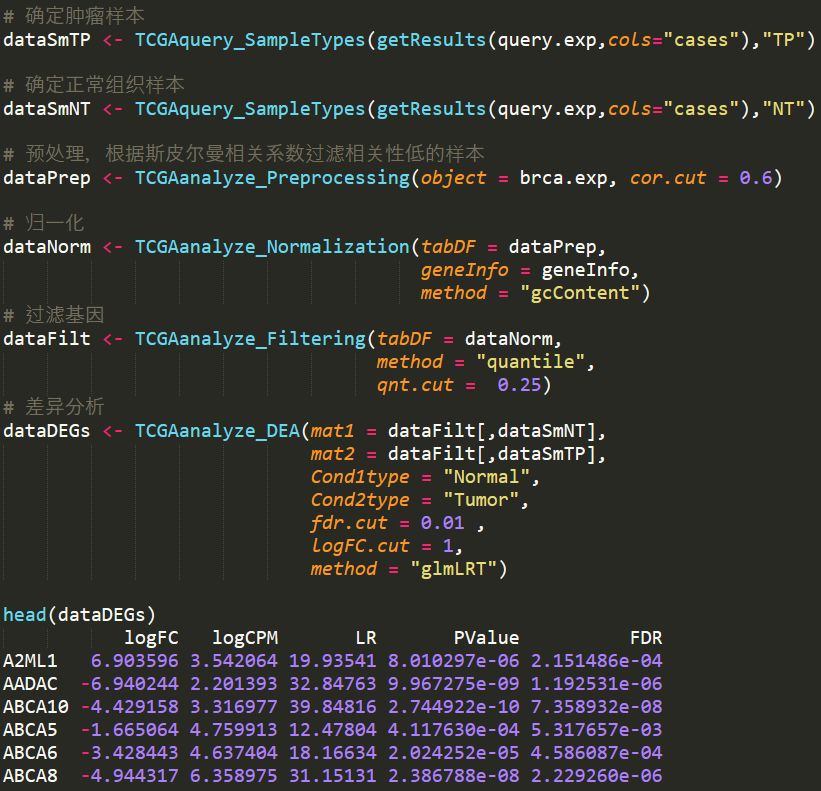
对数据进行预处理,根据样本间的斯皮尔曼相关系数去除相关性较低的样本
归一化,调用了EDASeq中的归一化算法
筛选基因,根据表达量的均值进行筛选
差异分析,调用了edgeR中的差异算法
代码如下
代码如下
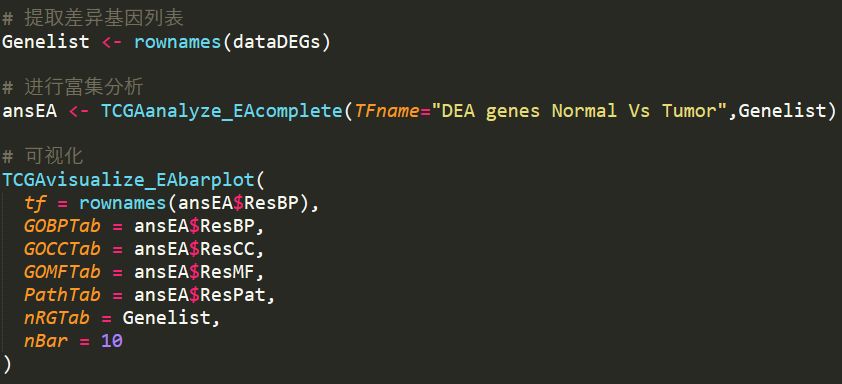
可视化的结果如下所示
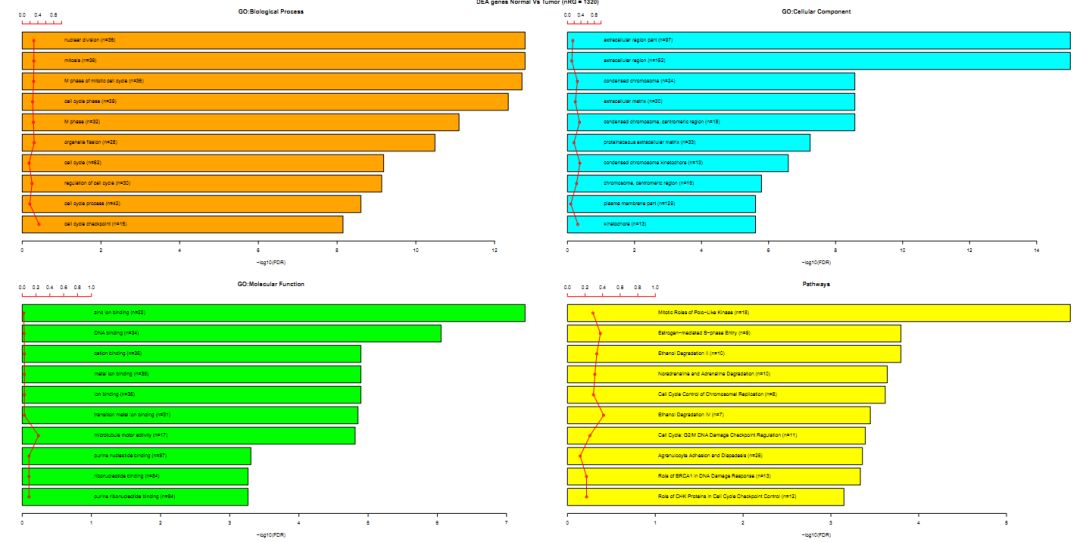
GO的3大类别加上kegg pathway共4个类别的数据,对应4张柱状图,每个柱状图展示的是FDR值最显著的top10个条目,横坐标我-log10(FDR), 散点代表的是GeneRatio, 也称之为enrich factor, 代表富集到该通路下的差异基因个数占该通路下所有基因总数的比例。
感谢各位的阅读,以上就是“如何使用TCGAbiolinks分析TCGA中的表达谱数据”的内容了,经过本文的学习后,相信大家对如何使用TCGAbiolinks分析TCGA中的表达谱数据这一问题有了更深刻的体会,具体使用情况还需要大家实践验证。这里是亿速云,小编将为大家推送更多相关知识点的文章,欢迎关注!
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。





