本篇文章为大家展示了怎么使用NetMHCpan进行肿瘤新抗原预测分析,内容简明扼要并且容易理解,绝对能使你眼前一亮,通过这篇文章的详细介绍希望你能有所收获。
NetMHCpan软件用于预测肽段与MHC I型分子的亲和性,最新版本为v4.0, 基于人工神经网络算法,以180000多个定量结合数据和MS衍生的MHC洗脱配体的组合为训练集构建模型。结合亲和力数据来自人,小鼠,猪等多个物种的MHC分子,MS洗脱的配体数据来自55个人和小鼠的HLA等位基因。
直接上传fasta格式的蛋白序列就可以了, 示意如下
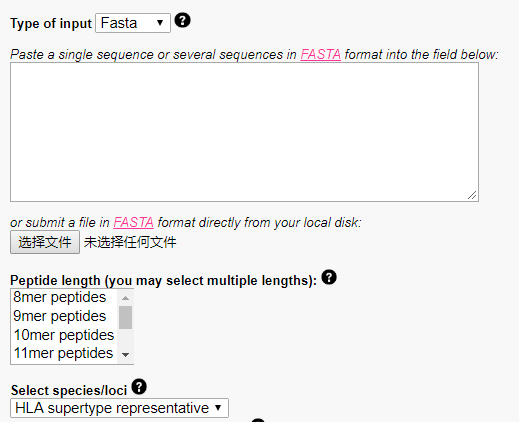
第一步上传涵盖了体细胞突变位点的氨基酸序列,上传的氨基酸序列是突变之后的序列,不是野生型的蛋白质序列。
第二步选择切割肽段的方式,抗原通过抗原表位与MHC分子结合,MHC I型分子可以结合的抗原表位长度为8到11个氨基酸,对应这里的8-11mer,先将蛋白质序列切分成短的肽段之后在进行MHC分子亲和性的预测。
第三步选择HLA allel, 确定之后点击提交按钮即可。输出结果示意如下
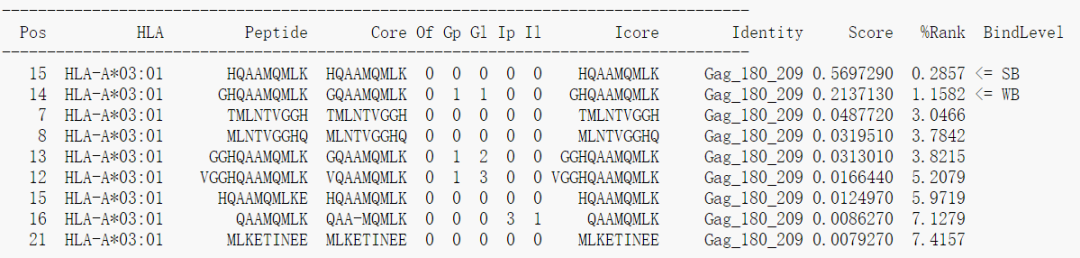
列数很多,其中的Peptide就是从原始的输入序列中提取出的长度为8-11个氨基酸的肽段,Pos对应肽段的在原始序列上的起始位置,第一个位置从0开始计数;Core对应与MHC结合的肽段序列,和blast类似,允许插入和缺失,%Rank代表该肽段是一个天然存在的肽段的可能性,数值越小越好,最后一列的BindLevel代表亲和力的强弱水平,SB表示strong binding, WB表示weak bingding。每一列的详细解释参见以下链接
http://www.cbs.dtu.dk/services/NetMHCpan/output.php
官方按照Rank值来筛选结果,默认情况下rank小于0.5的定义为强亲和性,rank值在0.5到2之间的定义为弱亲和性。通过该软件可以从突变之后的氨基酸序列中预测到与MHC I型分子亲和力较强的肽段,作为候选的肿瘤新抗原。
为了进一步简化分析,相关的数据分析pipeline被开发出来,只需要提供肿瘤患者的体细胞突变数据和HLA分型结果即可,软件自动提取突变氨基酸序列,并进行NetMHCpan分析,类似的软件有很多,NeoPredPipe软件就是其中之一,该软件的网址如下
https://github.com/MathOnco/NeoPredPipe
基本用法如下
python NeoPredPipe.py \-I somatic.vcf \-H hlatypes.txt \-o ./ \-n TestRun \-c 1 2 -E 8 9 10需要提供两个输入文件,-I指定体细胞突变的vcf文件,-H指定HLA分型结果文件。更多细节请参考该软件的官方文档。
通过上述的数据分析,可以快速定位出候选的新抗原,然而其中的假阳性率还是非常高的,后续还需要结合体外实验来进一步筛选和过滤。
上述内容就是怎么使用NetMHCpan进行肿瘤新抗原预测分析,你们学到知识或技能了吗?如果还想学到更多技能或者丰富自己的知识储备,欢迎关注亿速云行业资讯频道。
亿速云「云服务器」,即开即用、新一代英特尔至强铂金CPU、三副本存储NVMe SSD云盘,价格低至29元/月。点击查看>>
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。
原文链接:https://my.oschina.net/u/4580290/blog/4570396



 计算
计算 安全
安全 数据库
数据库 网络和加速
网络和加速 企业服务
企业服务



