peak calling软件MACS如何使用,针对这个问题,这篇文章详细介绍了相对应的分析和解答,希望可以帮助更多想解决这个问题的小伙伴找到更简单易行的方法。
MACS全称是Model-based Analysis of ChIP-Seq,是使用的最广泛的peak calling软件之一,其基本原理简介如下
在chip_seq等数据中科学家发现在真实的结合位点两侧,正负链的测序深度分布如下图所示,对应峰值的中心距离peak中心有一定的偏移
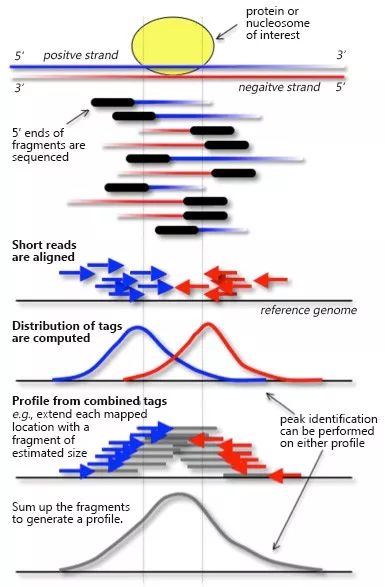
MACS首先通过一个模型来评估真实的peak中心和测序峰值的偏移距离,给定参数bandwidth和mfold, 采用一个大小为2倍bandwidth的滑动窗口,比较该窗口内真实测序深度的分布与随机测序的差异,如果二者的差异倍数超过了阈值mfold,则认为该窗口是一个peak区域。识别到初始的peak区域之后,随机挑选1000个高可信度的peak区域,分别计算正链和负链的测序深度分布,示意如下
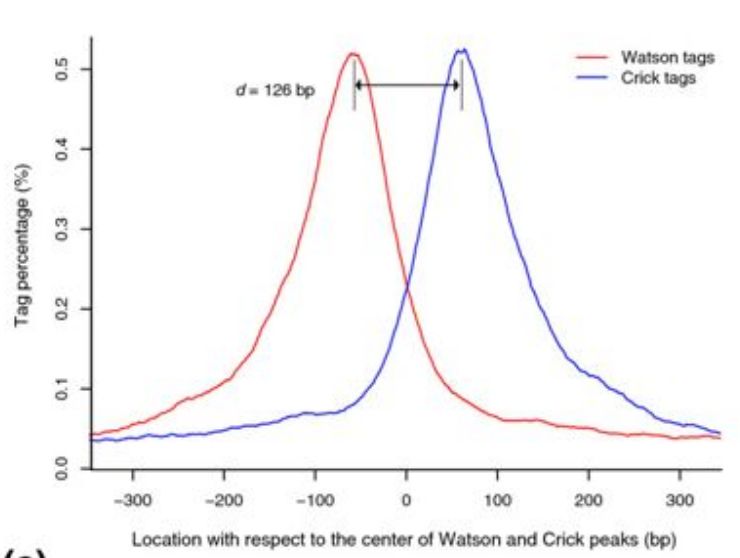
通过这种方式识别到正负链峰值之间的距离,定义为d。在后续peak calling时,会在初始计算结果的基础上向3’端偏移d/2的距离。
通常认为基因组上测序深度的分布是一个泊松分布,根据基因组测序深度的值,首先估算出整体泊松分布的参数
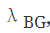
将初始peak区域偏移d/2的距离之后,再次已2d的滑动窗口进行滑动,寻找富集的区域,如果一个区域的测序分布的值大于整体泊松分布的值,则认为该区域是富集的。对于overlap的富集区域,进行合并,然后以peak中心区域开始,整体向3’端偏移d个碱基,其中测序深度最高的点作为peak的峰summit。
在比较input和IP样本的差异时,MACS也是基于泊松分布来计算的 ,只不过考虑到测序错误,重复区域,CNV等因素的影响,MACS采用了一个动态的泊松分布模型,即不是认为基因组所有区域都符合同一个泊松分布,而是认为不同的区域泊松分布的参数可以不同。对于候选的peak区域,通过以下模型去估算泊松分布的参数
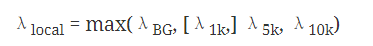
1k代表以peak中心为中心的1k窗口内的数据估算出来的值,后面的5k, 10k类似。然后通过这个局部泊松分布的参数去计算每个peak区域的p值,如果小于指定的pvalue的阈值,则输出该peak区域,该区域内IP样本的序列条数和局部泊松分布参数的比值作为该peak的fold enrichment。
关于peak calling软件MACS如何使用问题的解答就分享到这里了,希望以上内容可以对大家有一定的帮助,如果你还有很多疑惑没有解开,可以关注亿速云行业资讯频道了解更多相关知识。
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。





