еҰӮдҪ•иҝӣиЎҢMACS2 peak callingзҡ„е®һжҲҳ
д»ҠеӨ©е°ұи·ҹеӨ§е®¶иҒҠиҒҠжңүе…іеҰӮдҪ•иҝӣиЎҢMACS2 peak callingзҡ„е®һжҲҳпјҢеҸҜиғҪеҫҲеӨҡдәәйғҪдёҚеӨӘдәҶи§ЈпјҢдёәдәҶи®©еӨ§е®¶жӣҙеҠ дәҶи§ЈпјҢе°Ҹзј–з»ҷеӨ§е®¶жҖ»з»“дәҶд»ҘдёӢеҶ…е®№пјҢеёҢжңӣеӨ§е®¶ж №жҚ®иҝҷзҜҮж–Үз« еҸҜд»ҘжңүжүҖ收иҺ·гҖӮ
MACSжҳҜдёҖж¬ҫжңҖдёәжөҒиЎҢзҡ„peak callingиҪҜ件пјҢжңҖеҲқжҳҜй’ҲеҜ№иҪ¬еҪ•еӣ еӯҗзҡ„chipж•°жҚ®жқҘи®ҫи®Ўзҡ„пјҢеңЁжңҖж–°зүҲжң¬дёӯпјҢд№ҹж·»еҠ дәҶеҜ№з»„иӣӢзҷҪдҝ®йҘ°зҡ„йҖӮй…ҚгҖӮзӣ®еүҚжңҖж–°зүҲжң¬дёәv2.0,е®ҳзҪ‘еҰӮдёӢ
https://github.com/taoliu/MACS
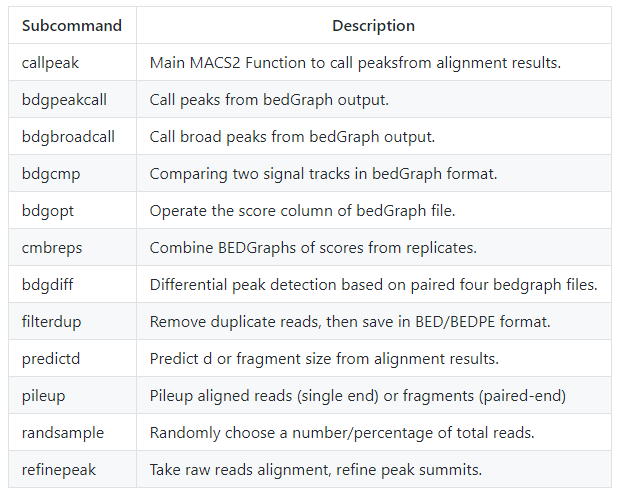
еңЁ2.0зүҲжң¬дёӯжҸҗдҫӣдәҶд»ҘдёӢеӨҡдёӘеӯҗе‘Ҫд»Ө
callpeak
bdgpeakcall
bdgbroadcall
bdgcmp
bdgopt
cmbreps
bdgdiff
filterdup
predictd
pileup
randsample
refinepeak
жҜҸдёӘеӯҗе‘Ҫд»Өе’ҢеҜ№еә”зҡ„еҠҹиғҪжҸҸиҝ°еҰӮдёӢ
дёӢйқўдё»иҰҒд»Ӣз»Қmacs2жңҖз»Ҹе…ёзҡ„дҪҝз”ЁеңәжҷҜpeak calling, еҹәжң¬з”Ёжі•еҰӮдёӢ
macs2 callpeak \
-t ip.bam \
-c input.bam \
--outdir out_dir \
-n chip \
-g hs
-tеҸӮж•°жҢҮе®ҡжҠ—дҪ“еӨ„зҗҶзҡ„ж ·жң¬пјҢ-cжҢҮе®ҡinputж ·жң¬пјҢеҖјеҫ—дёҖжҸҗзҡ„жҳҜпјҢmacsж”ҜжҢҒеӨҡз§Қж јејҸзҡ„иҫ“е…Ҙж–Ү件пјҢйҷӨдәҶдёҠиҝ°д»Јз ҒдёӯдҪҝз”Ёзҡ„bamж јејҸеӨ–пјҢиҝҳж”ҜжҢҒSAM/BEDж јејҸгҖӮ
--outdirжҢҮе®ҡиҫ“еҮәз»“жһңзҡ„зӣ®еҪ•пјҢ-nеҸӮж•°жҢҮе®ҡиҫ“еҮәж–Ү件еҗҚзҡ„еүҚзјҖпјҢ-gеҸӮж•°жҢҮе®ҡеҹәеӣ з»„зҡ„жңүж•ҲеӨ§е°ҸпјҢеңЁNGSж•°жҚ®дёӯпјҢжөӢеәҸreadsеңЁеҹәеӣ з»„дёҠзҡ„иҰҶзӣ–еәҰ并дёҚжҳҜ100%пјҢ иҖҢдё”жңүдәӣйҮҚеӨҚеҢәеҹҹзҡ„жҜ”еҜ№дҝЎжҒҜжҳҜдёҚеҸҜдҝЎзҡ„пјҢеү©дёӢзҡ„иғҪеӨҹеҲ©з”Ёзҡ„еҢәеҹҹйҖҡеёёеҸӘеҚ ж•ҙдёӘеҹәеӣ з»„еҢәеҹҹзҡ„70%еҲ°90%пјҢиҝҷдёӘеҢәеҹҹзҡ„еӨ§е°Ҹе°ұжҳҜжңүж•ҲеӨ§е°ҸпјҢеҜ№дәҺеёёи§Ғзҡ„зү©з§ҚпјҢзЁӢеәҸеҶ…зҪ®дәҶжңүж•ҲеӨ§е°ҸпјҢжҲ‘们еҸӘйңҖиҰҒжҢҮе®ҡзү©з§Қзҡ„зј©еҶҷеҚіеҸҜ

еҜ№дәҺе…¶д»–зү©з§ҚпјҢеҲҷйңҖиҰҒиҮӘе·ұжҢҮе®ҡжңүж•Ҳеҹәеӣ з»„зҡ„еӨ§е°ҸпјҢеҚ•дҪҚдёәbpгҖӮ
иҫ“еҮәж–Ү件еҰӮдёӢ
chip_model.r
chip_peaks.narrowPeak
chip_peaks.xls
chip_summits.bed
model.rжҳҜдёҖдёӘеҸҜжү§иЎҢзҡ„Rи„ҡжң¬пјҢйҖҡиҝҮд»ҘдёӢд»Јз ҒеҸҜд»Ҙдә§з”ҹдёҖдёӘPDFзҡ„иҫ“еҮәж–Ү件
Rscript chip_model.r
第дёҖйЎөиЎЁзӨәpeakйӮ»иҝ‘еҢәй—ҙжӯЈиҙҹй“ҫжөӢеәҸеҲҶеёғпјҢз”ЁдәҺиҜ„дј°dиҝҷдёӘеҸӮж•°еҖјпјҢзӨәж„ҸеҰӮдёӢ
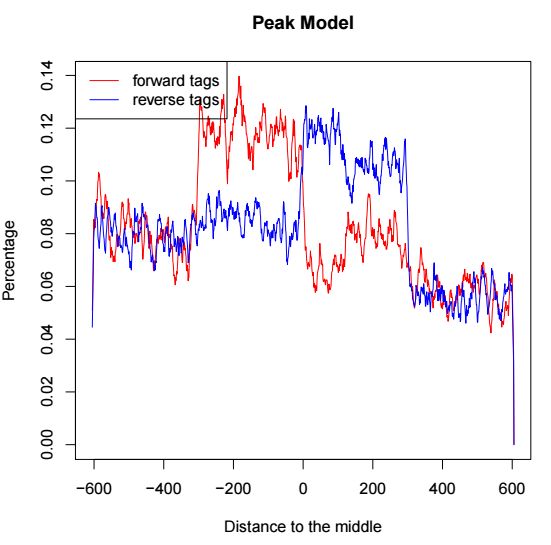
第дәҢйЎөжҳҜcross-correlationеҲҶжһҗзҡ„з»“жһңпјҢзӨәж„ҸеҰӮдёӢ

еҗҺзјҖдёәxlsзҡ„ж–Ү件жҳҜpeakзҡ„иҫ“еҮәз»“жһңпјҢеҶ…е®№зӨәж„ҸеҰӮдёӢ
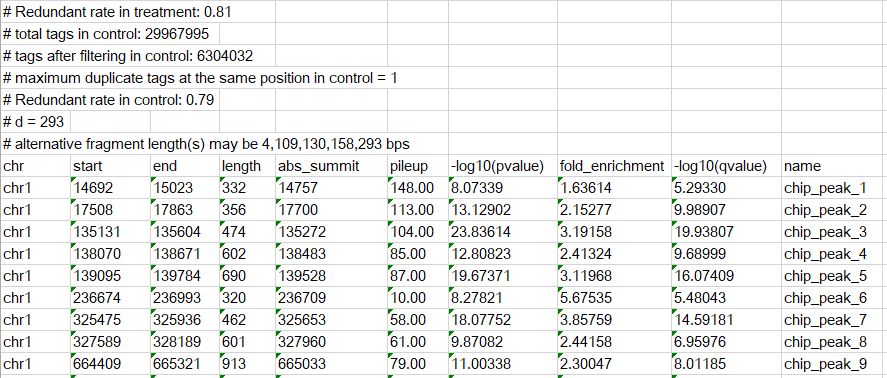
#ејҖеӨҙзҡ„жҳҜжіЁйҮҠдҝЎжҒҜпјҢжҳҫзӨәдәҶиҪҜ件и°ғз”Ёзҡ„е…·дҪ“е‘Ҫд»Өе’ҢеҸӮж•°и®ҫзҪ®пјҢдҫҝдәҺж ёжҹҘпјӣе…¶д»–зҡ„иЎҢи®°еҪ•дәҶpeakзҡ„еҢәй—ҙдҝЎжҒҜпјҢиҝҷйҮҢзҡ„иө·е§ӢдҪҚзҪ®йҮҮз”Ёзҡ„жҳҜд»Һ1ејҖе§Ӣи®Ўж•°зҡ„ж–№ејҸгҖӮ
еҗҺзјҖдёәnarrowpeakзҡ„ж–Ү件жҳҜдёҖдёӘBEDж јејҸзҡ„ж–Ү件пјҢеҶ…е®№зӨәж„ҸеҰӮдёӢ
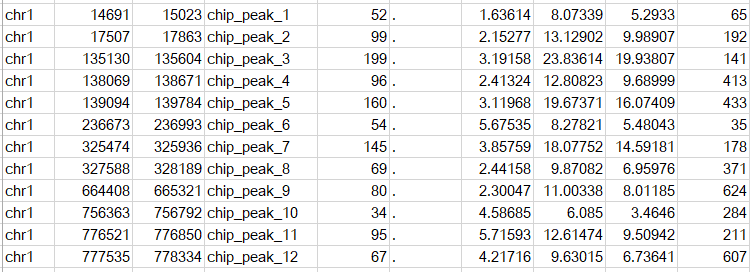
еүҚеӣӣеҲ—д»ЈиЎЁpeakеҢәй—ҙе’ҢеҗҚз§°пјҢжіЁж„Ҹbedж јејҸдёӯиө·е§ӢдҪҚзҪ®д»Һ0ејҖе§Ӣи®Ўж•°пјҢ第дә”еҲ—зҡ„еҖјдёәint(-10*log10qvalue)пјҢ第е…ӯеҲ—е…ЁйғЁдёә.,第дёғеҲ—дёәfold_enrichment,第八еҲ—дёә-log10(pvalue),第д№қеҲ—дёә-log10(qvalue),第еҚҒеҲ—дёәpeakзҡ„дёӯеҝғпјҢеҚіsummitи·қзҰ»peakиө·е§ӢдҪҚзҪ®зҡ„и·қзҰ»пјҢеҜ№еә”abs_summit - startгҖӮ
еҗҺзјҖдёәbedзҡ„ж–Ү件дёәpeakдёӯеҝғпјҢеҚіsummitеҜ№еә”зҡ„bedж–Ү件пјҢеҶ…е®№зӨәж„ҸеҰӮдёӢ
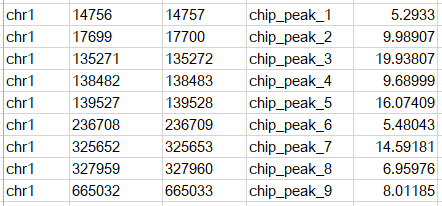
жңҖеҗҺдёҖеҲ—дёә-log10(qvalue)гҖӮ
зңӢе®ҢдёҠиҝ°еҶ…е®№пјҢдҪ 们еҜ№еҰӮдҪ•иҝӣиЎҢMACS2 peak callingзҡ„е®һжҲҳжңүиҝӣдёҖжӯҘзҡ„дәҶи§Јеҗ—пјҹеҰӮжһңиҝҳжғідәҶи§ЈжӣҙеӨҡзҹҘиҜҶжҲ–иҖ…зӣёе…іеҶ…е®№пјҢиҜ·е…іжіЁдәҝйҖҹдә‘иЎҢдёҡиө„и®Ҝйў‘йҒ“пјҢж„ҹи°ўеӨ§е®¶зҡ„ж”ҜжҢҒгҖӮ





