这篇文章主要介绍“如何使用python Genome Tracks可视化hi-c数据”,在日常操作中,相信很多人在如何使用python Genome Tracks可视化hi-c数据问题上存在疑惑,小编查阅了各式资料,整理出简单好用的操作方法,希望对大家解答”如何使用python Genome Tracks可视化hi-c数据”的疑惑有所帮助!接下来,请跟着小编一起来学习吧!
可视化是数据分析中非常重要的一个环节,对于NGS分析数据的可视化,最常用的就是各种基因组浏览器了,既有UCSC, GBrowse等基于web的基因组浏览器,也有igvtools等本地化的图形界面软件。对于Hi-C数据,在前面的文章中也介绍过基于web的WashU Epigenome Browser基因组浏览器和本地化的juicebox软件。
熟练掌握其中一个软件的用法就可以满足大部分的需求了,但是作为一个生信分析的极客,总感觉还是需要一款命令行工具来提高效率。python和R都拥有非常强大的可视化能力,今天介绍一款基于python语言的软件pyGenomeTracks, 一款原汁原味的命令行工具,拥有和基因组浏览器相同的展现形式,网址如下
https://github.com/deeptools/pyGenomeTracks
该软件支持可视化以下几种信息
bigwig
bed
bedgraph
links
Hi-C matrices
采用该软件可视化的效果图如下
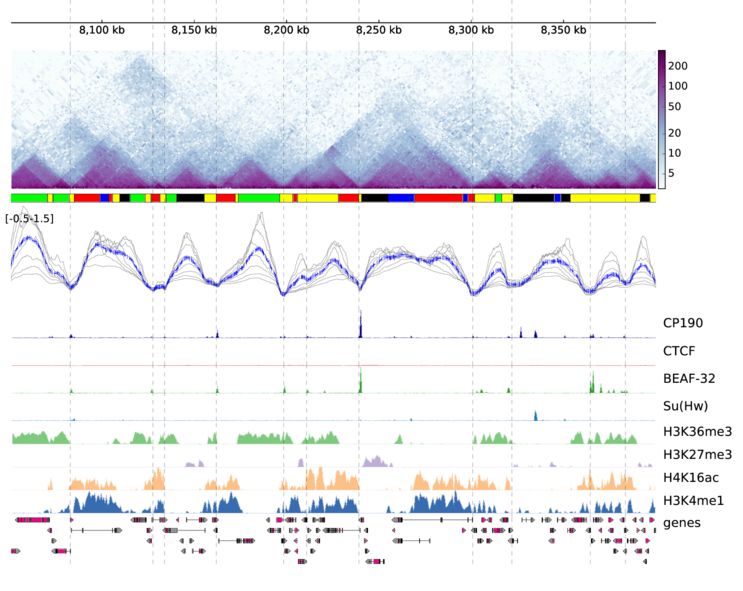
和基因组浏览器一样的展现形式,每一层为一个track。该软件采用配置文件的形式来配置需要展示的文件信息,每个需要展示的文件和对应的参数都写在一个标签下,具体写法如下
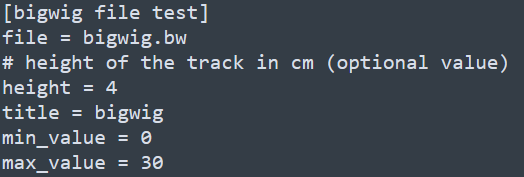
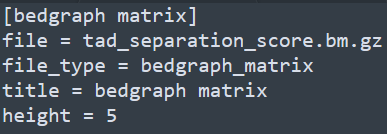
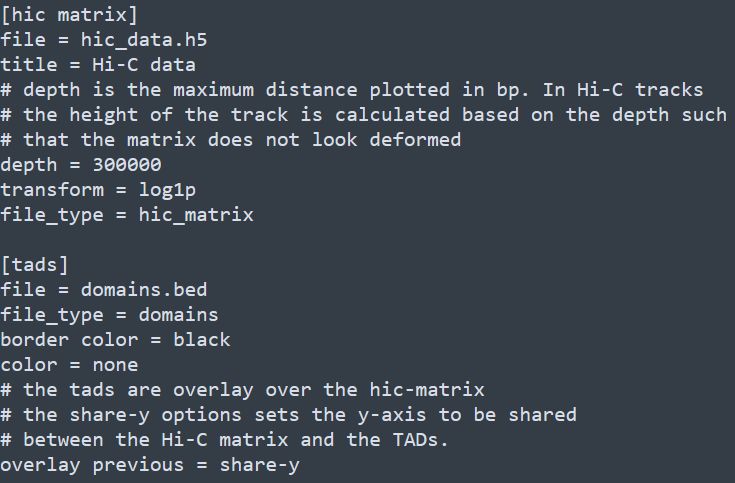
除此之后,还有x-axis和spacer等标签,分别对应x轴和两个tracks之间的空格区域。下方如下
[spacer][x-axis]where = top编辑好配置文件之后,就可以运行了,用法如下
pyGenomeTracks \--tracks tracks.ini \--region chr2:10,000,000-11,000,000 \--outFileName output.pdftracks参数指定配置文件的名称,region参数指定需要可视化的基因组区域,outFileName参数指定输出文件的名称。为了达到美观的效果,有许多的参数需要调整,更多细节请参考官方文档和示例。
一个hi-c数据可视化的效果图如下
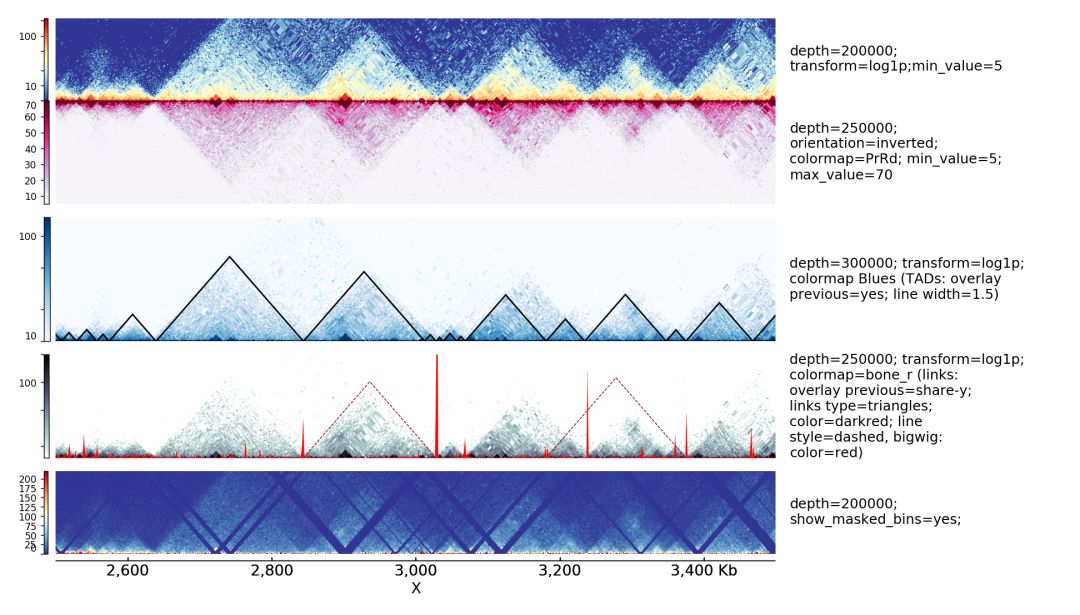
通过该软件,可以高效的展示hi-c数据。
到此,关于“如何使用python Genome Tracks可视化hi-c数据”的学习就结束了,希望能够解决大家的疑惑。理论与实践的搭配能更好的帮助大家学习,快去试试吧!若想继续学习更多相关知识,请继续关注亿速云网站,小编会继续努力为大家带来更多实用的文章!
亿速云「云服务器」,即开即用、新一代英特尔至强铂金CPU、三副本存储NVMe SSD云盘,价格低至29元/月。点击查看>>
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。
原文链接:https://my.oschina.net/u/4580290/blog/4570555



 计算
计算 安全
安全 数据库
数据库 网络和加速
网络和加速 企业服务
企业服务



