еҰӮдҪ•дҪҝз”ЁhomerиҝӣиЎҢpeakжіЁйҮҠ
жң¬зҜҮеҶ…е®№дё»иҰҒи®Іи§ЈвҖңеҰӮдҪ•дҪҝз”ЁhomerиҝӣиЎҢpeakжіЁйҮҠвҖқпјҢж„ҹе…ҙи¶Јзҡ„жңӢеҸӢдёҚеҰЁжқҘзңӢзңӢгҖӮжң¬ж–Үд»Ӣз»Қзҡ„ж–№жі•ж“ҚдҪңз®ҖеҚ•еҝ«жҚ·пјҢе®һз”ЁжҖ§ејәгҖӮдёӢйқўе°ұи®©е°Ҹзј–жқҘеёҰеӨ§е®¶еӯҰд№ вҖңеҰӮдҪ•дҪҝз”ЁhomerиҝӣиЎҢpeakжіЁйҮҠвҖқеҗ§!
homerиҪҜ件йӣҶжҲҗдәҶи®ёеӨҡзҡ„еҠҹиғҪпјҢеҢ…жӢ¬peak calling, peakжіЁйҮҠпјҢmotifеҲҶжһҗзӯүзӯүпјҢйҖҡиҝҮиҝҷдёҖдёӘиҪҜ件пјҢе°ұеҸҜд»Ҙе®ҢжҲҗchip_seqзҡ„з»қеӨ§йғЁеҲҶеҲҶжһҗеҶ…е®№пјҢдёҚеҸҜи°“дёҚејәеӨ§гҖӮжң¬ж–Үдё»иҰҒд»Ӣз»ҚиҝҷдёӘиҪҜ件иҝӣиЎҢpeakжіЁйҮҠзҡ„з”Ёжі•гҖӮ
еңЁhomerдёӯйҖҡиҝҮannotatePeaks.plиҝҷдёӘи„ҡжң¬иҝӣиЎҢpeakзҡ„жіЁйҮҠпјҢеҲҶдёәд»ҘдёӢдёӨжӯҘ
1. еҮҶеӨҮеҸӮиҖғеҹәеӣ з»„зҡ„жіЁйҮҠдҝЎжҒҜ
homerеҶ…зҪ®дәҶи®ёеӨҡзү©з§Қзҡ„жіЁйҮҠдҝЎжҒҜдҫӣжҲ‘们дёӢиҪҪпјҢйҖҡиҝҮд»ҘдёӢе‘Ҫд»ӨеҸҜд»ҘжҹҘзңӢжүҖжңүеҶ…зҪ®зҡ„зү©з§Қ
perl configureHomer.pl --list
е…¶дёӯGENOMESйғЁеҲҶеҜ№еә”зҡ„е°ұжҳҜеҶ…зҪ®ж”ҜжҢҒзҡ„зү©з§ҚпјҢйғЁеҲҶеҶ…е®№еұ•зӨәеҰӮдёӢ
GENOMES
v5.10 hg19 v6.0 human genome and annotation for UCSC hg19
+ mm10 v6.0 mouse genome and annotation for UCSC mm10
- sacCer3 v6.0 yeast genome and annotation for UCSC sacCer3
- panTro5 v6.0 human genome and annotation for UCSC panTro5
д»Ҙhg19дёәдҫӢпјҢдёӢиҪҪж–№ејҸеҰӮдёӢ
perl configureHomer.pl -install hg19
дёӢиҪҪзҡ„дҝЎжҒҜдҝқеӯҳеңЁhomerе®үиЈ…зӣ®еҪ•зҡ„dataзӣ®еҪ•дёӢпјҢд»Ҙhg19дёәдҫӢпјҢеңЁdata/genome/hg19зӣ®еҪ•дёӢпјҢж–Ү件еҲ—иЎЁеҰӮдёӢ
в”ңв”Җв”Җ chr1.fa
в”ңв”Җв”Җ chr2.fa
в”ңв”Җв”Җ chr3.fa
в”ңв”Җв”Җ ...fa
в”ңв”Җв”Җ chrom.sizes
в”ңв”Җв”Җ conservation
в”ңв”Җв”Җ hg19.annotation
в”ңв”Җв”Җ hg19.aug
в”ңв”Җв”Җ hg19.basic.annotation
в”ңв”Җв”Җ hg19.full.annotation
в”ңв”Җв”Җ hg19.miRNA
в”ңв”Җв”Җ hg19.repeats
в”ңв”Җв”Җ hg19.rna
в”ңв”Җв”Җ hg19.splice3p
в”ңв”Җв”Җ hg19.splice5p
в”ңв”Җв”Җ hg19.stop
в”ңв”Җв”Җ hg19.tss
в”ңв”Җв”Җ hg19.tts
в””в”Җв”Җ preparsed
еҢ…еҗ«дәҶеҸӮиҖғеҹәеӣ з»„зҡ„fastaеәҸеҲ—д»ҘеҸҠдёҚеҗҢеҢәеҹҹзҡ„еҢәй—ҙж–Ү件гҖӮ
hg19.basic.annotationеҶ…е®№еҰӮдёӢ
Intergenic chr1 1 10873 + N 1900000000
promoter-TSS (NR_046018) chr1 10874 11974 + P 1
non-coding (NR_046018, exon 1 of 3) chr1 11975 12227 + pseudo 125025
intron (NR_046018, intron 1 of 2) chr1 12228 12612 + I 810684
non-coding (NR_046018, exon 2 of 3) chr1 12613 12721 + pseudo 125026
intron (NR_046018, intron 2 of 2) chr1 12722 13220 + I 810684
non-coding (NR_046018, exon 3 of 3) chr1 13221 13361 + pseudo 125027
еҗҢж—¶еңЁdata/accessionзӣ®еҪ•дёӢпјҢиҝҳжңүеҸӮиҖғеҹәеӣ з»„еҜ№еә”зҡ„еҹәеӣ жіЁйҮҠж–Ү件гҖӮ
human2gene.tsvи®°еҪ•дәҶеҹәеӣ зҡ„ubigene id, gene symbolзӯүдҝЎжҒҜпјҢеҶ…е®№еҰӮдёӢжүҖзӨә
ADE73044 3107 Hs.656020 NM_002117 ENSG00000204525 HLA-C
ENSG00000113163 10087 Hs.270437 NM_005713 ENSG00000113163 COL4A3BP
DB065460 9947 Hs.132194 NM_005462 ENSG00000155495 MAGEC1
ENSP00000282466 285313 Hs.58561 NM_178822 ENSG00000152580 IGSF10
DB029361 22849 Hs.131683 NM_014912 ENSG00000107864 CPEB3
XP_016877211 87 Hs.235750 NM_001102 ENSG00000072110 ACTN1
EAW77897 56965 Hs.270244 NM_020213 ENSG00000137817 PARP6
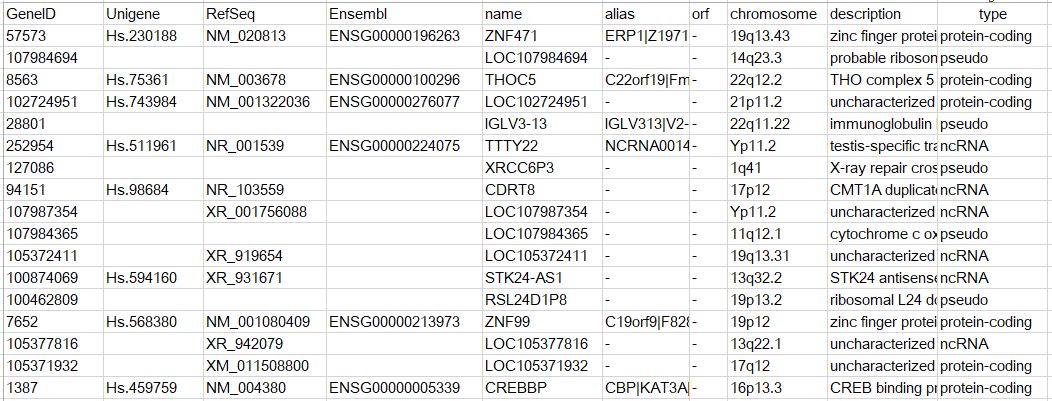
human.descriptionи®°еҪ•иЎЁдәҶеҹәеӣ зҡ„еҠҹиғҪжҸҸиҝ°пјҢзұ»еҲ«зӯүдҝЎжҒҜпјҢзӨәж„ҸеҰӮдёӢ
2. иҝӣиЎҢжіЁйҮҠ
з”Ёжі•еҰӮдёӢ
annotatePeaks.pl peak.bed hg19 > peak.annotation.xls
第дёҖдёӘеҸӮж•°дёәpeakзҡ„bedж–Ү件пјҢ第дәҢдёӘеҸӮж•°дёәеҸӮиҖғеҹәеӣ з»„зҡ„еҗҚз§°гҖӮиҫ“еҮәз»“жһңеҰӮдёӢжүҖзӨә
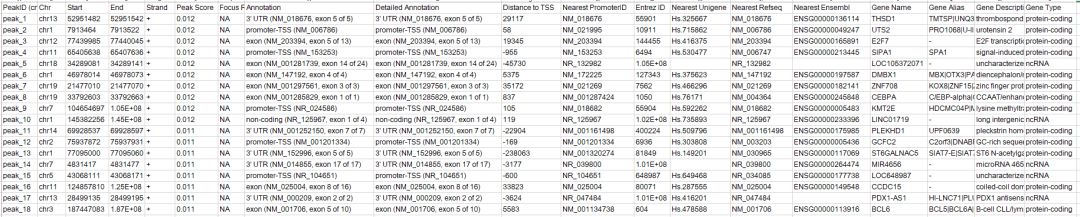
жіЁйҮҠзҡ„еҶ…е®№еҢ…еҗ«дёӨдёӘйғЁеҲҶпјҢ第дёҖйғЁеҲҶжҳҜи·қзҰ»peakеҢәй—ҙжңҖиҝ‘зҡ„иҪ¬еҪ•иө·е§ӢдҪҚзӮ№TSSпјҢ第дәҢйғЁеҲҶжҳҜеҜ№peakеңЁеҹәеӣ з»„еҢәеҹҹзҡ„еҲҶеёғпјҢжҜ”еҰӮTSS,TTS,3вҖҷUTR,5вҖҷUTRзӯүеҢәеҹҹгҖӮ
еҲ°жӯӨпјҢзӣёдҝЎеӨ§е®¶еҜ№вҖңеҰӮдҪ•дҪҝз”ЁhomerиҝӣиЎҢpeakжіЁйҮҠвҖқжңүдәҶжӣҙж·ұзҡ„дәҶи§ЈпјҢдёҚеҰЁжқҘе®һйҷ…ж“ҚдҪңдёҖз•Әеҗ§пјҒиҝҷйҮҢжҳҜдәҝйҖҹдә‘зҪ‘з«ҷпјҢжӣҙеӨҡзӣёе…іеҶ…е®№еҸҜд»Ҙиҝӣе…Ҙзӣёе…ійў‘йҒ“иҝӣиЎҢжҹҘиҜўпјҢе…іжіЁжҲ‘们пјҢ继з»ӯеӯҰд№ пјҒ





