这期内容当中小编将会给大家带来有关怎么使用tophat-fusion鉴定融合基因,文章内容丰富且以专业的角度为大家分析和叙述,阅读完这篇文章希望大家可以有所收获。
tophat-fusion 是一款利用RNA_seq 数据鉴定融合基因的工具。
该软件是集成在tophat软件中的,只需要安装好tophat之后就可以使用了,使用方法也比较简单,唯一需要注意的是目录结构。
tophat-fusion要求固定的目录结构,比如我在result文件夹下进行tophat-fusion的分析, 那么我需要在该目录下准备以下几个文件
物种对应的refGene.txt 和 ensGene.txt, 这两个文件可以从UCSC下载得到
新建一个blast 文件夹,注意文件夹的名字必须为”blast”, 在blast 文件夹下需要从NCBI下载nt, human_genomic, other_genomic开头的所有文件,下载的链接如下:
ftp://ftp.ncbi.nlm.nih.gov/blast/db/
结果输出目录,每个样本对应一个输出目录,输出目录的前缀为tophat_, 下划线之后加上样本名称,类似tophat_MCF,MCF为样本的名字
当然你还需要物种对应的bowtie1的索引文件,注意这里必须为bowtie1的索引, tophat检测融合基因时推荐bowtie1的索引方式。
上述文件都准备好之后,就可以开始分析了,步骤如下
第一步其实就是利用tophat将reads比对到参考基因组上,只不过对于融合基因的reads而言,其比对方式比较特殊,需要添加额外的参数,具体代码如下
tophat2 -o tophat_MCF7 -p 20 --fusion-search --keep-fasta-order --bowtie1 --no-coverage-search -r 0 --mate-std-dev 80 --max-intron-length 100000 --fusion-min-dist 100000 --fusion-anchor-length 13 --fusion-ignore-chromosomes chrM hg19_bowtie1/hg19 SRR064286_1.fastq SRR064286_2.fastq
在result目录下,直接运行如下代码就可以了
tophat-fusion-post -p 20 --num-fusion-reads 1 --num-fusion-pairs 2 --num-fusion-both 5 hg19_bowtie1/hg19
默认处理的是human的融合基因,如果是其他物种,需要添加--non-human参数。
tophat-fusion会根据目录结构自动识别对应的样本, 运行完成之后,会生成一个名为tophatfusion_out的文件夹,该文件夹下是所有样本的融合基因分析结果。
我们只需要看其中的result.html文件就可以了,内容示意如下
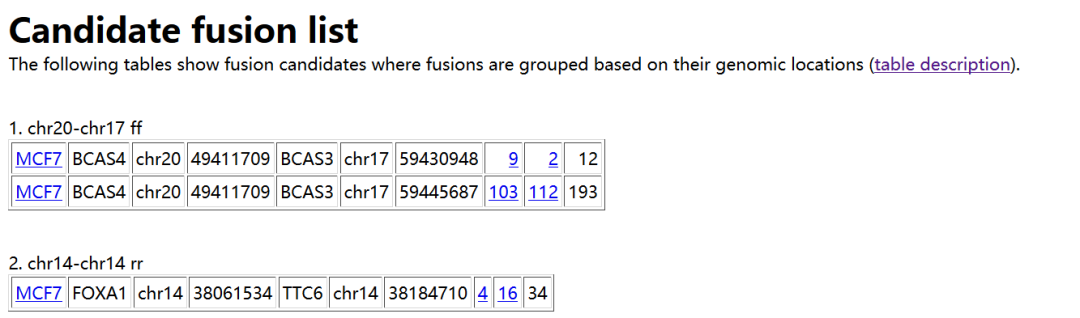
每一列的含义如下
Sample name in which a fusion is identified
Gene on the “left” side of the fusion
Chromosome ID on the left
Coordinates on the left
Gene on the “right” side
Chromosome ID on the right
Coordinates on the right
Number of spanning reads
Number of spanning mate pairs
Number of spanning mate pairs where one end spans a fusion
相比fusionmap, 该软件的运行时间特别的长。
上述就是小编为大家分享的怎么使用tophat-fusion鉴定融合基因了,如果刚好有类似的疑惑,不妨参照上述分析进行理解。如果想知道更多相关知识,欢迎关注亿速云行业资讯频道。
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。





